日付;2022/08/22(月)
日付;2025/09/09(火)ngs_disambiguateについて追記
これまでにWindows 11のWindows subsystems for Linux(wsl、バージョン2; wsl2)にUbuntu(20.04)をインストールしたので、 今回はRNA-seqの解析で、特にリードのフィルタリング、リファレンスシークエンス、マッピング、いわゆるプリプロセッシングというヤツに主に使うソフトをインストールしたいと思う。これらは既にwsl2にインストールされたDebian(Debian 9)で動作を確認しているので、ubuntuでも当然動くと思う。今後使っていく上でもしなにか不都合がみつかったら、その都度この記事も訂正してこうと思う。
なんと、STARとsamtools以外は全部バイナリを使うという始末である。でも、自分はBioinformaticianじゃあないし、それでいい。動けば良い。正直、こんなもんは技術員レベルの仕事とみなされる可能性があり、イキがれるほどのモノでもないし。Linux使えれば誰だって簡単にできるし。みんなが使うエクセルやワードと変わらん。バイナリでOK。
必要なパッケージのインストール
sudo apt update
sudo apt search build-essential
sudo apt install build-essential
sudo apt install zip
sudo apt install defalt-jre ## trimmomaticで必要。
sudo apt install gedit ## Ubuntuの標準テキストエディタはNanoというらしいが、万人が使いやすいようにするためのLinuxディストリビューションなのになぜこんなガチテキストエディタなんだよ。
sudo apt install zlib1g-dev ## STARで必要。
sudo apt install libncurses5-dev ## samtoolsで必要。
sudo apt install liblzma-dev ## samtoolsで必要。
sudo apt install libbz2-dev ## samtoolsで必要。
sudo apt install libcurl4-nss-dev ## samtoolsで必要。
sudo apt install git Anaconda
wget https://repo.anaconda.com/archive/Anaconda3-2022.05-Linux-x86_64.sh
sh Anaconda3-2022.05-Linux-x86_64.shPython 2.7の環境を設定する
後にインストールするfusionCatcherとrseqcはpython 3.9が使えないので、python 2.7の環境をセットアップしておく必要がある。
conda create -n py27 python=2.7 anaconda
conda activate py27 ## これでpython 2.7の環境ができる。これを走らせると、プロンプトがpy27になる。
conda deactivate ## python 2.7を使い終わったらこれで終了させる必要がある。FastQC
wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.9.zip
unzip fastqc_v0.11.9.zipTrimmomatic
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
unzip Trimmomatic-0.39.zip
java -jar trimmomatic-0.39.jarパスを通しても”Unable to access jar file ./trimmomatic-0.39.jar”とか言われる場合があるが、そのときは特に何も考えずに、trimmomatic-0.39.jarが置いてあるところまでcdで移動して”java -jar trimmomatic-0.39.jar”か”java -jar ./trimmomatic-0.39.jar”を打って実行する。これで動くんだから、これで良い。
PRINSEQ++
wget https://github.com/Adrian-Cantu/PRINSEQ-plus-plus/releases/download/v1.2.4/binary_prinseq-plus-plus-1.2.4.tar.gz
tar -zxvf binary_prinseq-plus-plus-1.2.4.tar.gzマニュアルにはconda installでインストールできると書いてあるが、conda installでもconda install -c biocondaでもインストールできなかった。
STAR
まずは最新のソースをダウンロードする。
wget https://github.com/alexdobin/STAR/archive/2.7.10a.tar.gz
tar -xzf 2.7.10a.tar.gz
cd STAR-2.7.10a次にコンパイルする。
cd STAR/source
make STARsamtools
以下はマニュアル通りのインストールなのだが、途中にある”./configure –prefix=/home/XXXXX/samtools”というヤツ、どうも動いていない気がする。もしかしたら./configureだけで良いかもしれない。
wget https://github.com/samtools/samtools/releases/download/1.16/samtools-1.16.tar.bz2
tar -jxvf samtools-1.16.tar.bz2
cd samtools-1.16
./configure --prefix=/home/XXXXX/samtools
make
make installngs_disambiduate
これは任意の配列を標的とするFASTQファイルから分けるライブラリであるが、これ同じことがSTARとかでも出来る。でも、当時はけっこう便利だった。ここ(https://github.com/AstraZeneca-NGS/disambiguate)にある。以下はbiocondaにあるPre-compile versionである。
conda install -c bioconda ngs-disambiguate
source .bashrc
ngs_disambiguaterseqc
これも古いPythonでしか動かない。幸いpython 3.0未満では動くらしい(rseqc -> python[version=’3.4.*|3.5.*|3.6.*|<3.0.0′]と出力された。)ので、fusionCatcherと同じ用にpython 2.7の環境で動かす。
conda activate py27
conda install -c bioconda rseqc
/home/XXXXX/anaconda3/envs/py27/bin/bam2fq.py -h
conda deactivatefusionCathcer
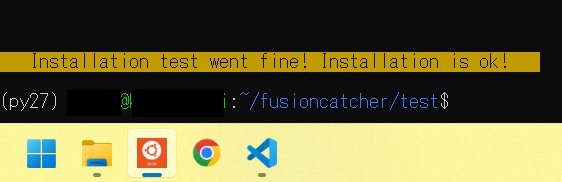
python 2.7の環境でしか動かないし、メモリも最低で24GB必要である。これらがなければインストールできない。マニュアルにはストレージに700GBの空きが必要とか書いてあるが、どうやらそれがなくても動くようだ。
インストール中にヒトのシークエンスをダウンロードするところがあるが、合計16GB以上ダウンロードするので、場合によってはかなり時間がかかる。また、最後のインストールのテストでもあまり遅いCPUだと時間がかかるかもしれない。
conda activate py27
wget http://sf.net/projects/fusioncatcher/files/bootstrap.py -O bootstrap.py && python bootstrap.py -t --download #最後のデータベースのダウンロードは時間がかかるので要注意。
cd /home/XXXX/fusioncatcher/test
./test.sh
conda deactivate個人的に要注意なのは、インストール中にThreadsを何個使うか聞かれるところがある。それを読み飛ばして「Y」とか入力してしまうと、1 Threadを使う設定になってしまい、最後のテストさえもなかなか終わらない、なんてことになるので、そこはちゃんとコンピューターをフル活用できるような数を入れたほうが良い。この場合はちゃんと「n」にして、次に12を入力した。
このインストールは、fusioncatcherが使うソフトのインストールをいちいち「Y」と入力して行く必要がある。ちょっと面倒だが、まぁ自分はbootstrap.pyをイジる自信がないので、しょうがなく確認しながら「Y」を入力していった。
BWA
これらはfastq_screenで必要。
wget https://sourceforge.net/projects/bio-bwa/files/bwa-0.7.17.tar.bz2
tar -jxvf bwa-0.7.17.tar.bz2
cd bwa-0.7.17]
sudo makeBowtie
これらはfastq_screenで必要。
wget https://sourceforge.net/projects/bowtie-bio/files/bowtie/1.3.1/bowtie-1.3.1-linux-x86_64.zip
unzip bowtie-1.3.1-linux-x86_64.zipBowtie2
これらはfastq_screenで必要。
wget https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.4.5/bowtie2-2.4.5-linux-x86_64.zip
unzip bowtie2-2.4.5-linux-x86_64.zipfastq screen
wget https://github.com/StevenWingett/FastQ-Screen/archive/refs/tags/v0.15.2.tar.gz
tar -xvzf v0.15.2.tar.gz
cd /home/katsu/FastQ-Screen-0.15.2次にBWAでもBowtie2でもBowtieでも、解析で使用するレファレンスシークエンスでインデックスファイルを作る。
## building idex using BWA
time bwa index /home/kats/fastq_screen_working_dir/GRCh38_2022_06_06/GRCh38.primary_assembly.genome.fa
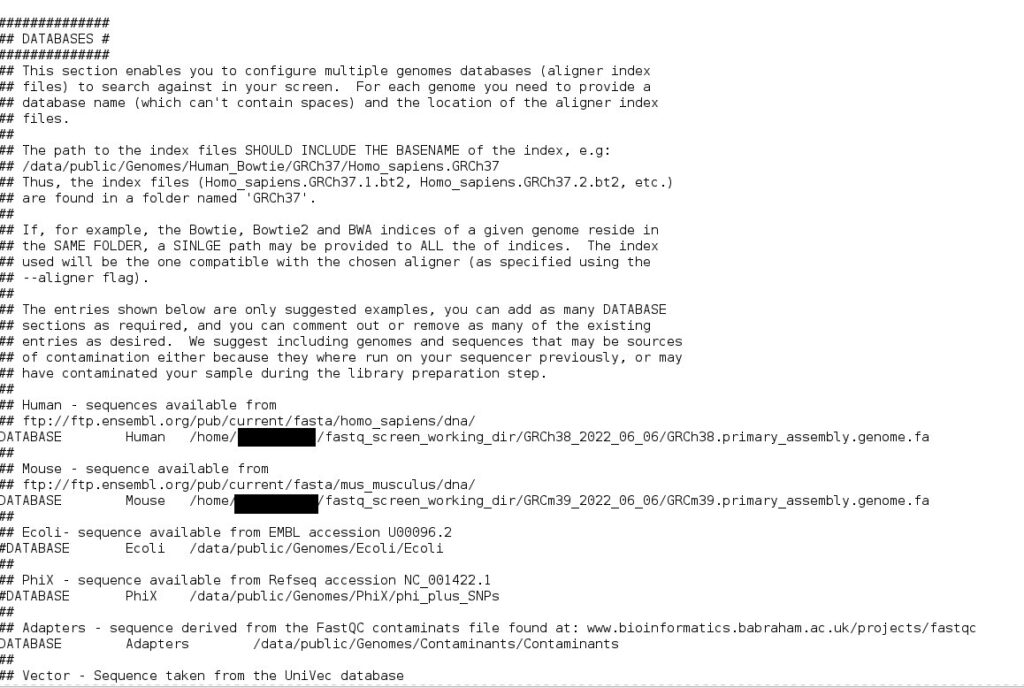
time bwa index /home/kats/fastq_screen_working_dir/GRCm39_2022_06_06/GRCm39.primary_assembly.genome.fa次に設定ファイルにBWA、Bowtie、Bowtie2、生物種毎にインデックスしたファイルのあるパスを追加(コメントアウトの”#”を消して、正確な完全パスに書き直す。)する。
cp fastq_screen.conf.example fastq_screen.conf
gedit fastq_screen.conf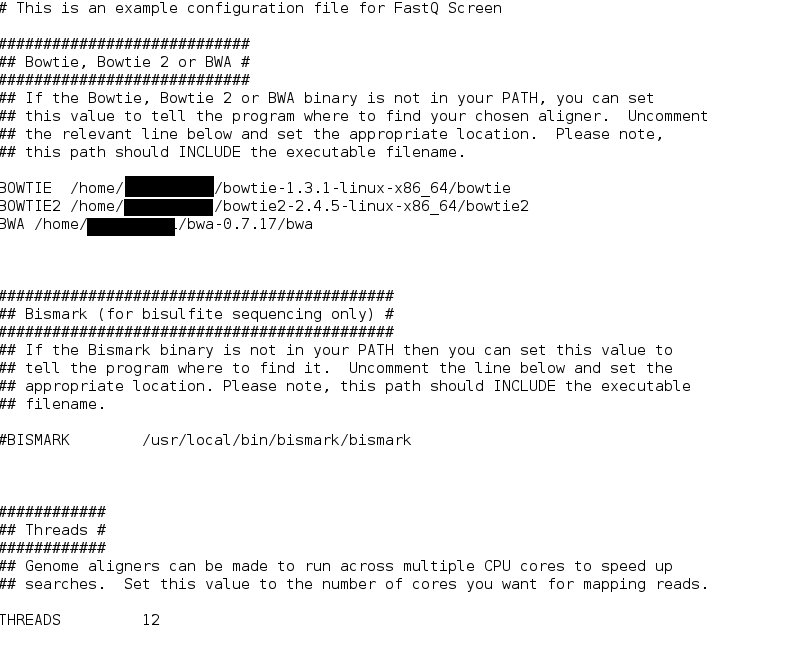
パスを通す
これも忘れずに。cdでホームディレクトリに移動して、.bashrcにPATH=$PATH:パスを加える。次にsource .bashrcで読み込む。これやらないと、いちいちバイナリが置いてあるディレクトリまで移動してからそれを使うハメになる。それは流石にめんどくさい。
cd
ls -a
gedit .bashrc
#以下の様にバイナリへのパスを記述して保存して閉じる。
PATH=$PATH:/home/XXXX/FastQC