日付;2022/09/19(月)、2022/10/02(日)改定と追記、2024/03/31(日)有料化
ゲノム解析で遊ぼうと思っても、肝心なデータがない。そういう場合は、NCBI(National Center for biotechnology Information)のSRA(Sequence Read Achive)からダウンロードして使用する。
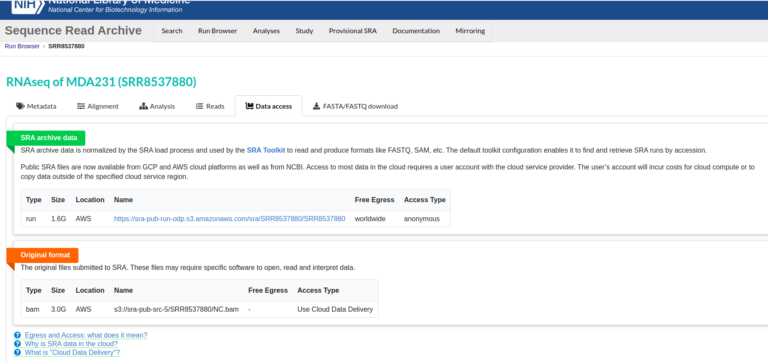
しかし、2021年くらいからNCBIが一部データをon-premisesで管理するのを止めて、AWSやGCPに保存するようになった(https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud/)。新しいデータ、例えばCOVID-19のシークエンスリードなんかがAWSやGCPに保存されているようだ(https://www.ncbi.nlm.nih.gov/sra/docs/sra-aws-download/)。
何が嫌だって、これは場合によってはダウンロードにお金がかかる場合がある。このsra-toolsの機能をフル活用するためには、AWSのEC2インスタンスとS3にサインアップしないといけない。言うても、かなりの量のダウンロードや長時間インスタンスを走らせなければ大金が請求されることはないのだろう。しかし、シークエンサーで読んだデータはかなり大きいので、注意しなくてはならないだろう。長時間のダウンロードならばEC2のランニングでもお金を取られる可能性もある。本当にこれらのデータが必要な場合は、職場からお金を支払ってもらうなどの対処が必要になりそうだ。これはSRA toolkitのインストラクションにも書いてあった。しかし、フリーと言っておきながら金がかかる場合があるってのは、どうも腑に落ちないところだ。
そういうことで、それが発表されて以来、 sra-toolsを使うのを、インストールでされ敬遠してきた。しかし、現職場でこの手の解析を行うようになってからは、どうしてもこれを使ってテストしたりしなければならなくなった。なので、今回はこの sra-toolsを使ってみようと思う。そのインストール時のメモをここに記しておこうと思う。
ポイントとしては「ちゃんとマニュアルを読んだ」ってところだ。金がかかるかもしれないとか、本当に嫌だ。結論としては、特に金がかかることはなさそうだった。ただ自分の勉強不足で怯えていただけらしい。