日付;2022/10/22(土)、2024/03/31(日)有料化、2024/06/08(土)Leading edge analysisについて追記
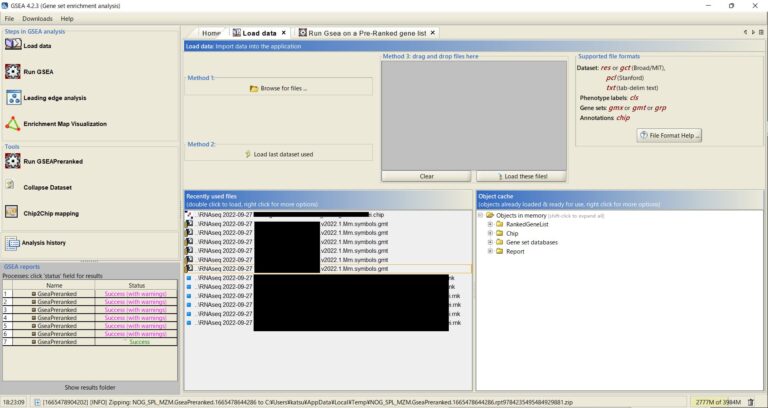
先日、Single Sample(N=1)のRNA-seqは意味があるのか無駄なのかを書いたが、そうは言っても、いろいろな事情によりそのような解析が必要な場合もある。それに、うまく行けばのサンプルの遺伝子発現プロファイルからそのサンプルがどのような細胞や組織に属する推測できるかもしれない。「最終的にお蔵入りになるから無駄だ」とか言っている自分も、結局のところその解析が必要だった。それに、この手の解析、特にGSEA(Gene Set Enrichment Analysis)の手順なんか、いちいち忘れてしまう愚かしい頭脳の持ち主である。正直こういうことを浅はかな知識で公開するのは良いことなのかわからないが、覚書くらいにはなるだろうと思ったので記すことにする。これはSinlge Sample(N=1)でGSEAをやる方法である。ちなみに、既にソフトのインストールは済んでいるものとして記述する。インストールは簡単なので大丈夫だろう。さすがに。
複数サンプルのGSEAは別の記事にまとめた
ここでは、サンプルが一つだけでも実施可能なGSEA(Single Sample GSEA; ssGSEA)について述べた。もし複数サンプルのGSEAを行いたい場合は、このリンクの記事を参考にすると良い(https://kts-research-blog.com/2024/05/27/gsea%e3%81%ae%e6%89%8b%e9%a0%86%e3%81%a8%e7%b5%90%e6%9e%9c%e3%81%ae%e8%a6%8b%e6%96%b9/)。結果までの一連の手順を書いた。この記事ではPreranked GSEA(すなわちsingle sample GSEA)の手順のみを書いたが、上記のリンクではLeading edge analysisからEnrichmentMapでのネットワークの描画まで書いた。Leading edge analysisのところはPreranked GSEAでも参考になると思う。
GSEAのマニュアル
マニュアルはここ。各種ファイルフォーマットについてはここ。変な解析をやってしまう前に、一度これは読んだ方が良いと思う。しかし、いちばん重要な結果の見方がしっかり書かれていないように思うのは自分だけだろうか。いずれにせよ、読んでいて損はないと思う。
GSEAのダウンロード
ここからダウンロードする。ダウンロードするためには自分のメールアドレスだのを登録する必要がある。WindowsとMac版などがあるが、個人的に自分のコンピュータに合わせれば良いと思う。コマンドラインもある。Rのバージョンもあるが、2019版(2022年10月の時点)らしいので、もしかしたら少し注意が必要な可能性もある。自分はR版は使ったことがないので、よくわからない。というか、2019年版のヤツなんて、あんまり使いたくない。