日付;2024/06/23(日)、2025/04/03(木)ssGSEA2に使用するGCTファイルについて追記
はじめに
これまでにBroad instituteのGSEAによりGSEAを行う方法をそれぞれ複数サンプルからなる2群間比較の場合と1群のみの場合について書いたが、ここではRを使ってsingle sample GSEA(ssGSEA)する方法を述べる。ちなみに、以前書いたBroad instituteのGSEA Prerankedを使った解析はN数を1とか2しか採っていなかった、もしくは1群しかないような、一体なんの目的でRNAシークエンスを行ったのかよくわからない、言わばしゃあなしで行う場合だった。これはライブラリやソフトウェアの取り扱い易さに関わる違いでしかないかもしれないが、ここで述べる方法はそのようなしゃあなしのGSEA RPrerankedとは目的が異なる。例えは、臨床サンプルが数十例あり、各々のサンプルを、ある遺伝子セットのエンリッチメントスコアで分類するような場合である。確かに、Broad instituteのGSEA Prerankedでもほぼ同じことが出来る。しかし、あれは1検体ずつ、もしくは、1群の代表値を使って解析を行うGUIベースのソフトであり、そんなことを数十例にわたって行うってのは、正気の沙汰では無い。
そんな時はRなどが強いと思う。以下に、どんな場合にどれを使うか(というか、自分ならばどうするか)について表にした。
| ライブラリもしくはソフト | 用途 | ウェブサイト |
|---|---|---|
| GSEA (Broad institute) | 2群間の比較であり、かつ、各群のサンプル数がそれなりに多い(少なくともn =3)場合。 | https://www.gsea-msigdb.org/gsea/index.jsp |
| GSEA Preranked (Broad Insitute) | 1群のみで、群間の比較ができない場合。 | https://www.gsea-msigdb.org/gsea/index.jsp |
| fgsea, GSVA, escape, ssGSEA2 | single sampleレベルでGSEAが可能であるため群の数やサンプルの数は問わない。しかし、既にGSEAを群間で行っており、対照群にくらべて目的の群においてある遺伝子セットが有意にupregulationもしくはdownregulationしている場合、その遺伝子セットがどのサンプルでどの程度upregulation/downregulationしているのか明らかにする場合に有効。そうでなければ、上記のGSEA prerankedと似たようなことをすることになる。single cell RNA-sequenceにも適応可能である。 | fgsea(https://bioconductor.org/packages/release/bioc/html/fgsea.html) GSVA(https://bioconductor.org/packages/release/bioc/html/GSVA.html) escape(https://bioconductor.org/packages/release/bioc/html/escape.html) ssGSEA2(https://github.com/nicolerg/ssGSEA2) |
Broad InstituteのGSEAの使い方やその結果の見方を学んでいる者であれば、以下のライブラリを使って得た結果を比較的簡単に理解できると思う。ここではその結果の見方や解釈は述べずに、Rによるそれらの使用方法を中心に記す。
Single sample GSEAを行う目的とそのコツ
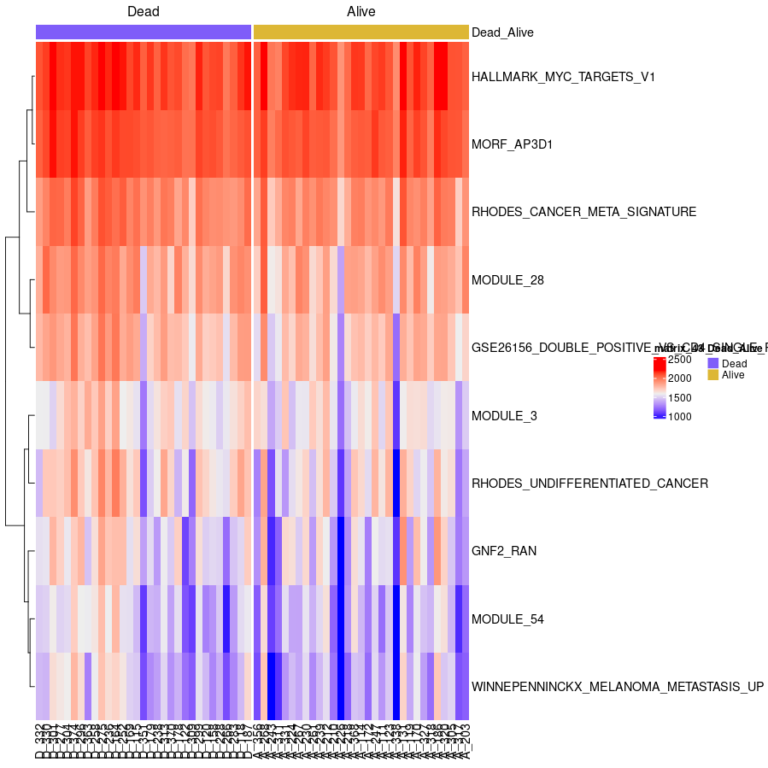
この解析を行う際のコツは、どの遺伝子セットに着目するか決めてから行うことであると考えている。Broad InstituteのGSEAは、基本的にはそれぞれ複数のサンプルを持つ2群を比較する方法を採っている。すなわち、2群間を比較することでどの遺伝子セットがどの程度エンリッチされているか求め、それらの違いをPermutation testで検定するという手法を採る。しかし、single sample GSEA(ssGESA)は群間の比較と言うより、各サンプル毎にGSEAを行うことで、どのサンプルで目的の遺伝子セットがエンリッチされているのか評価するところに焦点があるように思う。つまり、MsigDBに登録されている遺伝子セット全部を使って解析し、そこから着目する群でUpregulateもしくはDownregulateしている遺伝子セットを見つけ出すため、ということでは無く、対照群と比較して統計的有意にUpregulate/Downregulateしている遺伝子セットを既にいくつか見つけており、その次の解析として、それらの遺伝子セットは個々のサンプルでどんな具合になっているかを確認する、もしくは、上述したように個々のサンプルを分類するため等の目的で使用する、という具合である。シングルセルRNAシークエンス(scRNA-seq)でescapeを使用したことのある人ならわかると思うが、MsigDBに登録されている遺伝子セットを全部使って、初っ端から個々の細胞をescapeにより分類しようとしても相当数のエンリッチメントスコアが出てきてしまうのでそれは無謀である。次元削減を行うのは当然として、cellcypistやsingleRなどで各クラスターがどの細胞を示しているのかの当たりをつけ、さらに列をクラスター、行を各遺伝子の発現量から計算したランク(平均値、中央値、その他、もっと賢そうな値などの代表値で良いと思う)で構成されるマトリックスに落とし込んで、ようやくここで述べるライブラリ(fgsea、GSVA、escape、ssGSEA2等)でその細胞タイピングの結果の裏を取る、という感じで解析を行わなければ、露頭に迷うことになる。同定したクラスターの裏付けや、その細胞群に関連する遺伝子セットの探索などに使用するのが、single sample GSEAやsingle cell GSEAと理解している。
ここに記したfgsea、GSVA、escape、ssGSEA2は、しかしながら、おそらくssGSEA2は時間がかかるだろうが、scRNA-seqにも適用できる。以下にRのコードを順次述べる。