2026/01/03(土);初稿
2026/01/12(月);追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う。
- 1 はじめに
- 2 注意点1
- 3 注意点2
- 4 GDC portalからTCGAのカウント値をダウンロードする
- 5 使用するパッケージ
- 6 RNA-seqのカウントデータを読み込む
- 7 ここまでのRのダウンロード(文章を読みたくない人用)1
- 8 カウントデータと臨床情報を関連付ける
- 9 Anndataオブジェクトを作成(Optional)
- 10 ここまでのRのダウンロード(文章を読みたくない人用)2
- 11 escapeとGSVAを用いたエンリッチメント解析
- 12 ヒストグラムとバイオリンプロットによる疾患毎の遺伝子量またはエンリッチメントスコアの比較
- 13 カプランマイヤー法による生存率の解析
- 14 解析環境を記録しておく
- 15 まとめ
- 16 ここまでのRのダウンロード(文章を読みたくない人用)3
- 17 2026/01/12(月)追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う
はじめに
以前、TCGAのデータを使って、がんや腫瘍における遺伝子発現量(RNA-seqのカウント値)と患者の生存率の関係をRで解析したが、そのときは対応する正常組織(おそらくがんや腫瘍周辺の正常部位)におけるRNA-seqのカウン値をダウンロードしなかったため、それらをバイオリンプロットを用いた疾患毎の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアの比較に含めることが出来なかった。それに、TCGAのデータも2024年のバージョン41であり、ちょっと時間が経っていたので、改めてダウンロードし、解析することにした。
早速本題に入ろうと思う。
注意点1
以前の解析との違いは、以下である。
- 正常組織の遺伝子発現量を解析、特にバイオリンプロットに含めた。
- GDC portalからTCGA(とCPTAC)で採られたRNA-seqのカウント値をダウンロードした。
- ssGSEA2とfgseaは計算が遅すぎたので使用しなかった。escapeとGSVAのみでGene Set Enrichment Analysis(GSEA)を行った。
- MSigDBに登録されている(2025年10月時点)ヒトの全遺伝子セットを使ってGSEAした。
ここでやっている解析は以前の解析とほぼ同じであり、もしかしたら以前の解析の方が良いかもしれない。特に、後ほどバイオリンプロットで各疾患毎さらに対応する正常組織の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアを比較するが、正直、結論を得るためには正常組織の症例数が足りないように思うし、当然ながら正常組織を採取できない症例や疾患もあるためだ。正常組織も解析に含めてみて、これに気がついた。今なら、「正常組織も解析に含めることは出来ますが、症例数がちょっと足りないようにも思いますし、そもそも正常組織を採っていない疾患も多いです。もしかしたら、正常組織との比較は参考程度にしかならず、それで何かを決定するのは微妙かもしれません。」と自信を持って言える。
なお、CPTACについては今後、CPTACで採られたタンパク質発現量と遺伝子発現量の相関を解析するためである。ここではデータのクリーニングのときにちょっとだけ出てくるが、この記事ではそれらを解析しない。以下に比較表を載せておく。
| 項目 | この記事 | 以前の記事 |
|---|---|---|
| タイトル | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.43, 2025) | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.41, 2024) |
| TCGAのバージョン | v.43, 2025 | v.41, 2024 |
| GDC portalからのデータのダウンロード方法 | あり | なし |
| GMTファイルの作り方 | なし | あり(ただし、Geneontologyから選んできた遺伝子セット1つだけ) |
| 使ったGMTファイル | MsigDBに登録されているヒトの遺伝子セット全部(Human MSigDB v2025.1.Hs、2025年10月時点) | Geneontologyから選んできて自分で作成したHRR(Homologous Recombination Repair)と、MsigDBのH: hallmark gene sets(Human MSigDB v2025.1.Hs、2025年10月時点)のみ。 |
| エンリッチメント解析 | GSVA escape | ssGSEA2 escape GVSA fgsea |
| バイオリンプロットによる疾患毎の遺伝子発現量もしくは遺伝子セットのエンリッチメントの比較 | 正常組織あり ウィルコクソンの順位和検定あり(ただし、正常組織の例数が腫瘍組織に比べて少ないのでちょっと注意が必要と思う。) | 常組織なし ウィルコクソンの順位和検定なし |
| 生存期間解析でのログランク検定 | あり | あり |
| その他 | 比較するデータの数(リストの要素の数)が多く、そのために少しデータの準備がすこし複雑でメモリを喰いまくる。 | 比較するデータの数(リストの要素の数)がそんなに多くないので、データの準備がそんなに大変じゃない。 |
注意点2
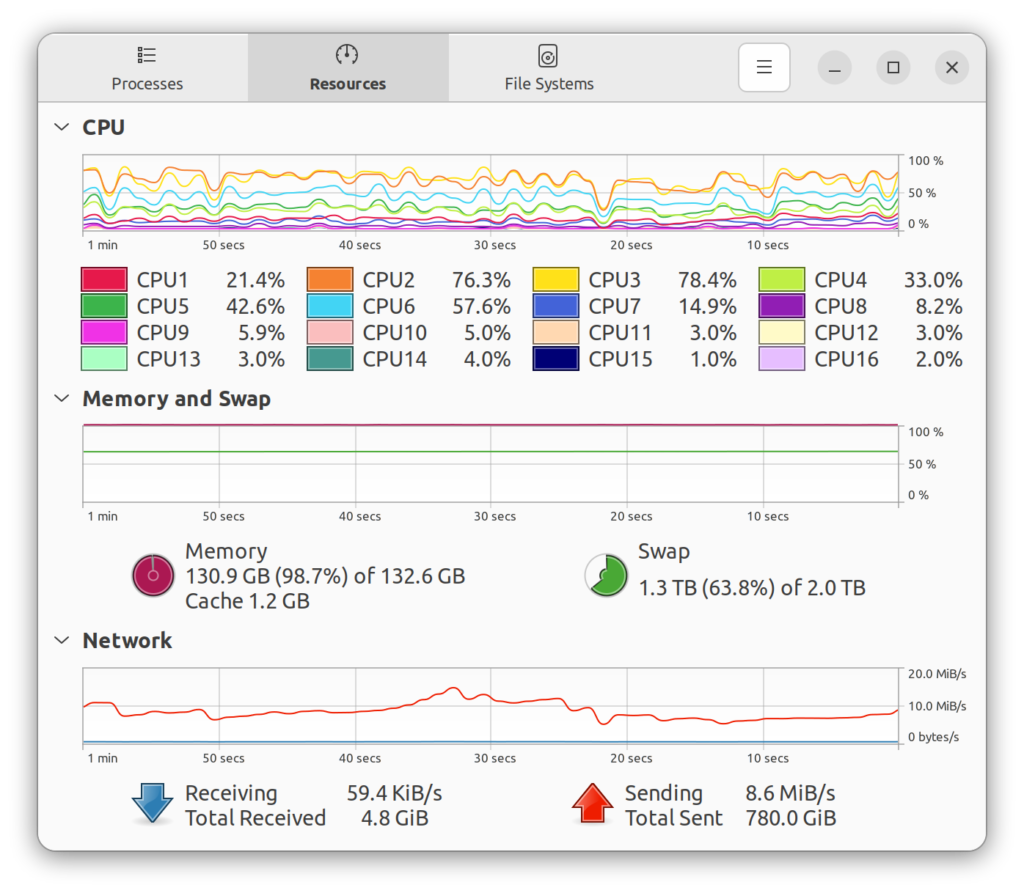
以前の解析でも書いたが、この解析で使用するコンピューターの性能はずいぶんと高いと思う。以下の画像の通りメモリを合計で1.3TB以上も使っており、たとえRandom Access Memory(RAM)(物理メモリ)を256GB積んでいたとしても全く足りていないことがわかる。ちなみに、これはデータセットの全部がすべて整って、生存率曲線の解析とそれに続くログランク検定を行っているとき(3つめのRのコード)のメモリの使用量である。1つ目から3つ目のコードを一つのRStudioに入れようとしても、作業領域が一杯になってまともに動かなくなってしまう。また、昨今のノートPCや軽量のラップトップでは計算能力が足りない可能性があるので、要注意である。もはやサーバーのようなコンピューターが必要と言える。
GDC portalからTCGAのカウント値をダウンロードする
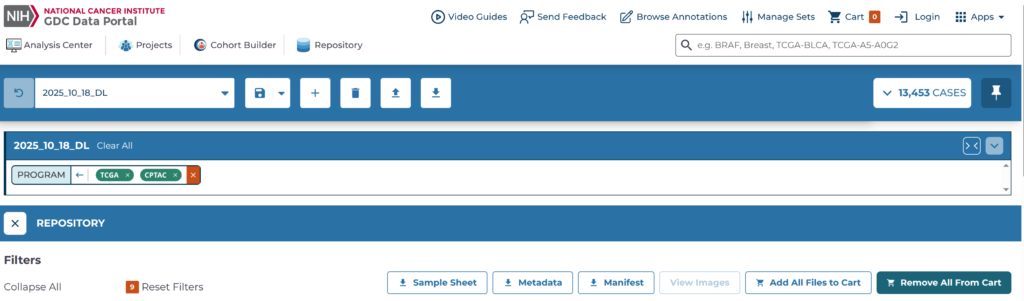
まずは、GDC portal(https://portal.gdc.cancer.gov/)からTCGAとCPTACのRNA-seqのカウント値をダウンロードする。ダウンロード方法については以前の記事が詳しいので、そちらを参照したほうが良いかもしれない。GDC portalは以下のようなフロントエンド(2025年12月時点)である。まず「Cohort Builder」を押す。
「Cohort Builder」を押すと、以下のようなページに入る。「Program」というパネルには様々なプロジェクト(の大元)があるので、TCGAとCPTACにチェックを入れて、そのコホートに名前を付けて保存する。こうすれば一々TCGAとCPTACを選び直して…ということが無くなる。ここでは2025_10_18_DLという名前で保存した。次に、「Repository」をクリックする。
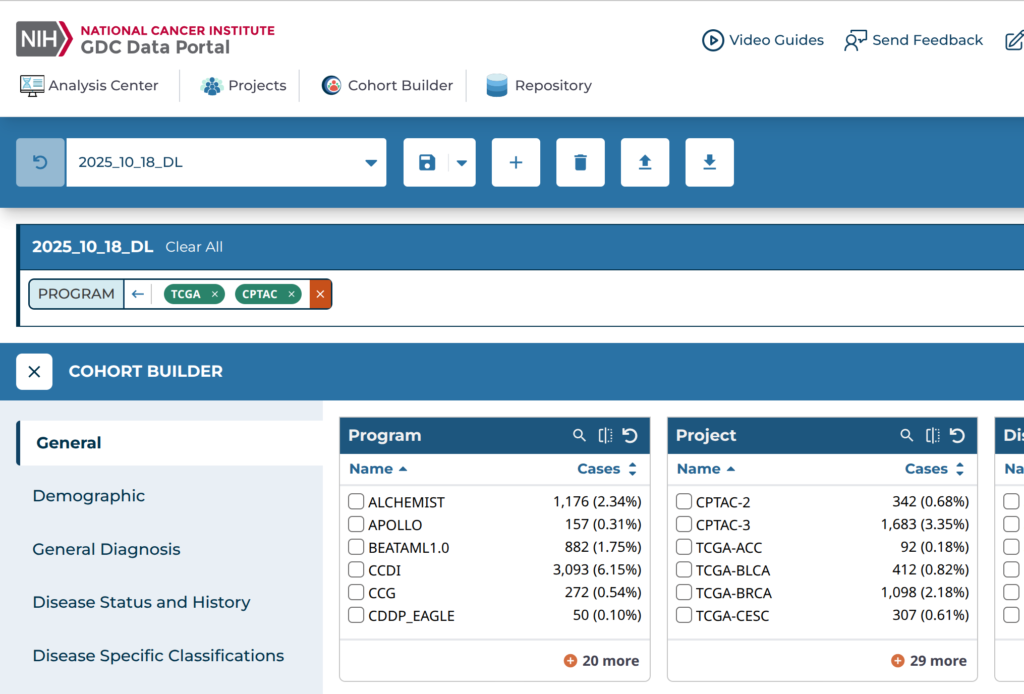
| Cohort Builder | 値 |
|---|---|
| Program | TCGA CPTAC |
次に「Repository」をクリックし、Filterのパネルのところを、以下の表のようにチェックを入れていく。なお、ダウンロードしたい症例はCartに入れてからまとめてmanifest fileなどをダウンロードするが、このとき、Cartには10,000ファイルしか入らなかったと思うので、以下のように順番1から3のように分割して、Cartに入れるファイル数を10,000より少なくしてからダウンロードすることにした。
ダウンロード1回目
まずは1回目のダウンロードが以下の表である。
| Filter | 値 | 順番 |
|---|---|---|
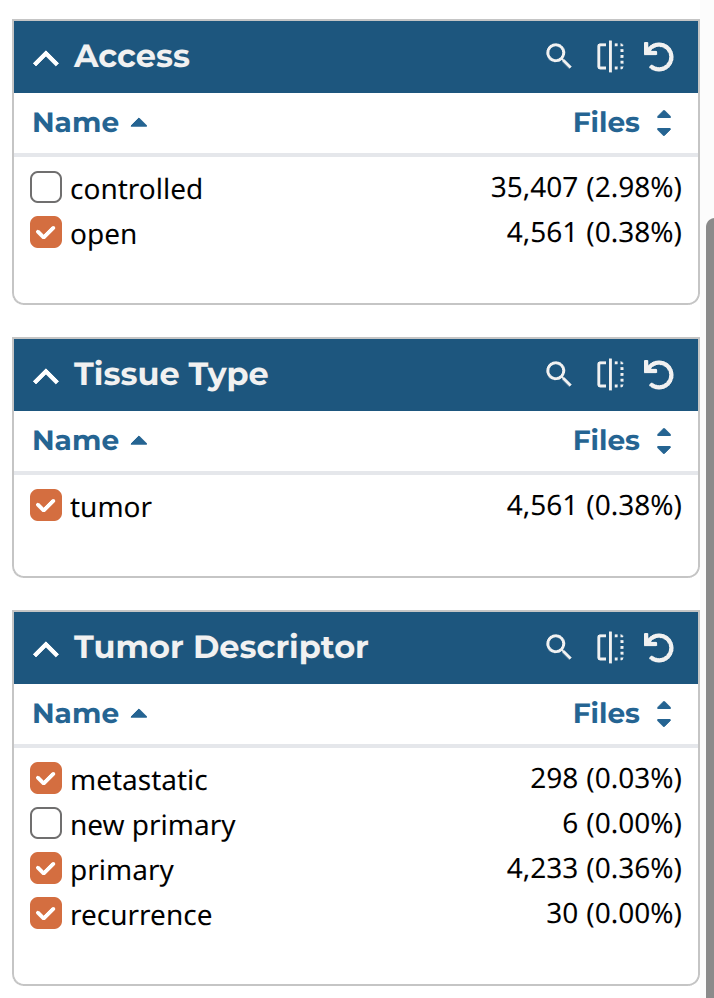
| Access | open | 1 |
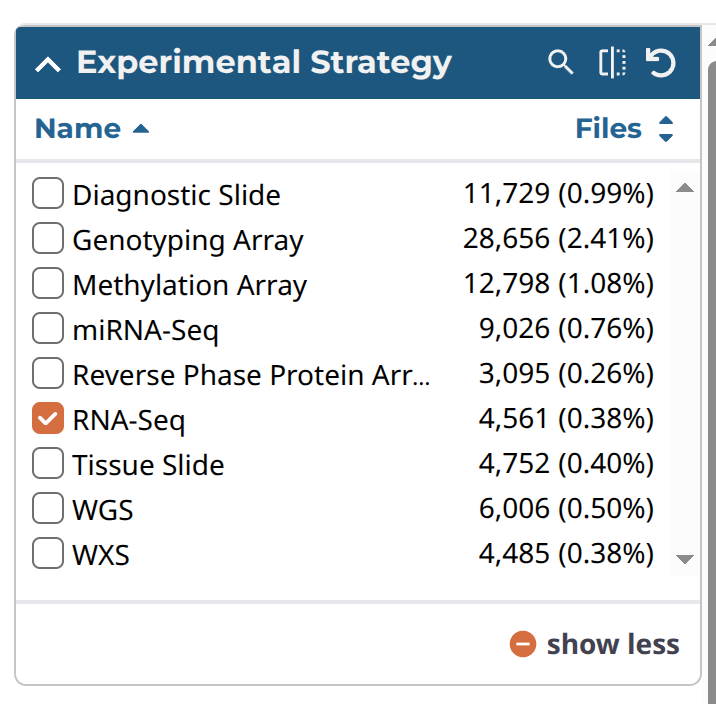
| Experimental Strategy | RNA-seq | 1 |
| Tissue type | tumor | 1 |
| Tissue Descriptor | metastatic primary recurrence | 1 |
| Preserved Method | ffpe frozen oct | 1 |
各パネルの画像は以下のような感じである。
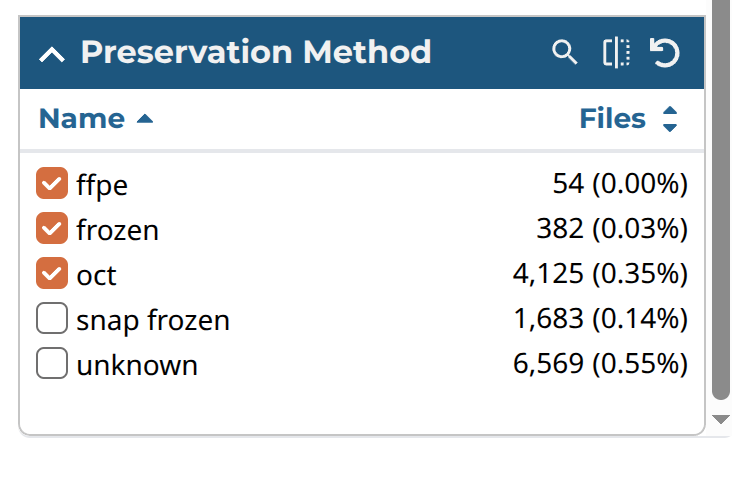

上記を選んだら、「Add All Files to Cart」をクリックし、次に右上にある「Cart」をクリックする。
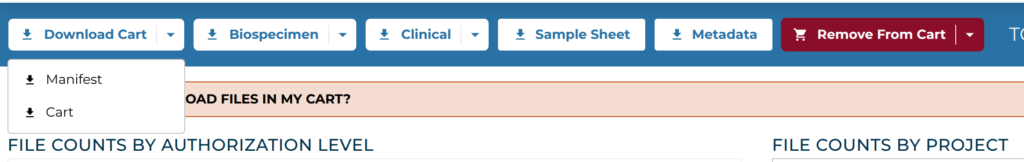
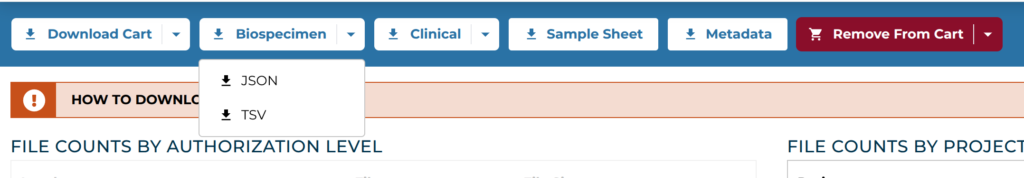
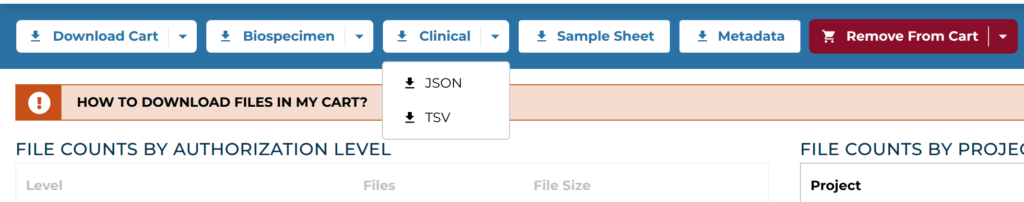
カートに移動したら、Manifestファイル、サンプルに関するデータ(Biospecimen)、臨床情報に関するデータ(Clinical)、Sample Sheet、Metadataをダウンロードする。Sample SheetとMetadataは表示の「Sample Sheet」、「Metadata」のところをクリックするだけである。他は以下のようにダウンロードする。BiospecimenとClinicalに関するファイルは圧縮されているので、適宜展開する。
ダウンロード2回目
以下がダウンロード2回目の値である。対象となるファイル選んでカートにいれたら、カートに移動して手順1の通り各種ファイルをダウンロードする。
| Filter | 値 | 順番 |
|---|---|---|
| Access | open | 2 |
| Experimental Strategy | RNA-seq | 2 |
| Tissue type | tumor | 2 |
| Tissue Descriptor | metastatic primary recurrence | 2 |
| Preserved Method | snap frozen unkown | 2 |
ダウンロード3回目
以下がダウンロード3回目の値である。3回目は正常組織のみを選んだ。これは、後ほどバイオリンプロットにより疾患間の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアを比較するときに、正常組織の発現量も加えるためである。手順1と2と同様に、対象となるファイル選んでカートにいれたら、カートに移動してダウンロード1回目の通り各種ファイルをダウンロードする。
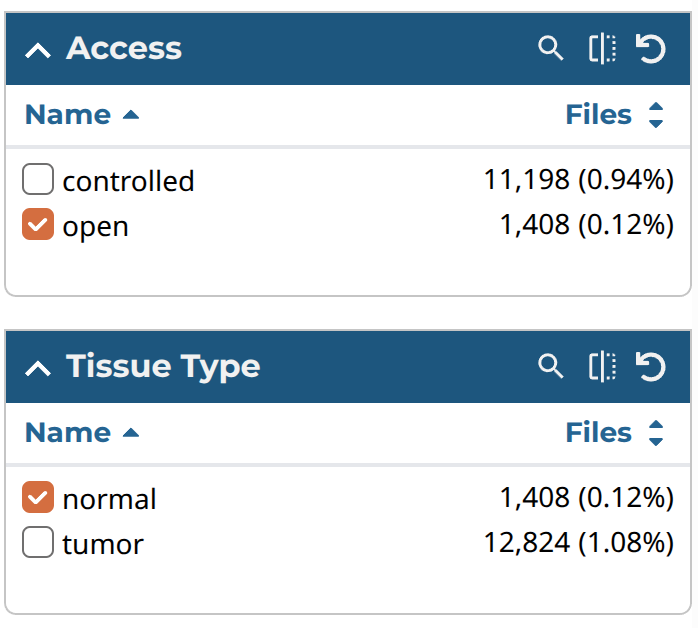
| Filter | 値 | 順番 |
|---|---|---|
| Access | open | 3 |
| Experimental Strategy | RNA-seq | 3 |
| Tissue type | normal | 3 |

gdc-clientを使ってManifest fileに記載されたファイルをダウンロードする
以下のコードがManifest fileを使ったRNA-seqのカウント値のダウンロード方法である。gdc-clientをここからダウンロードし、インストールする必要がある。これらの方法は以前の記事にある程度詳しく書いてある。以下がgdc-clientを使ったダウンロードの方法である。
# 1
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest.2025-10-25.154055.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest_1_log.txt"
# 2
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_manifest.2025-10-25.160033.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_manifest_2_log.txt"
# 3
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest.2025-10-25.160847.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest_3_log.txt"
# 3 retry
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest.2025-10-26.024514.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest_3_retry_log.txt"
# 1 retry
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest.2025-10-26.103837.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest_1_retry_log.txt"
# 2 retry
/mnt/team4tb/Dropbox/Downloads Safari/gdc_manifest.2025-10-27.003414.txt
gdc-client download -m "/mnt/team4tb/Dropbox/Downloads Safari/gdc_manifest.2025-10-27.003414.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_manifest_2_retry_log.txt"
使用するパッケージ
以下のパッケージが自分がいつもRNA-seqの解析に使っているものである。TCGAのデータを使った生存率解析のためのパッケージとして必須なものは、tidyverse、data.table、suvival、survminer、 grid.Extraである。それに加えて、この解析ではescapeとgsvaを用いる。
# DEG
library(baySeq)
library(edgeR)
library(DESeq2)
library(limma)
# enrichment analysis
library(clusterProfiler)
library(fgsea)
library(ssGSEA2) # devtools::install_github("nicolerg/ssGSEA2") # devtools::install_github(broadinstitute/ssGSEA2.0) and devtools::install_github(broadinstitute/ssGSEA2) did not work.
library(escape)
library(GSVA)
library(pathview)
# immune cell deconvolution
library(ConsensusTME)
library(estimate)
library(immunedeconv)
library(MCPcounter)
library(msigdbr)
library(quantiseqr)
library(TIMER) #devtools::install_github('hanfeisun/TIMER'); it will not work!! use TIMER of immunodeconv if you need.
library(xCell)
# stats
library(NSM3) # sudo apt install libgmp-dev libgmp10 libgmp3-dev
library(pwr)
library(missForest)
# basic
library(beeswarm)
library(circlize)
library(org.Hs.eg.db)
library(org.Mm.eg.db)
library(doParallel)
library(foreach)
library(parallel) # Need it for use of baySeq
library(pheatmap)
library(rtracklayer)
library(rvest) # this is for extraction of html table; results of leading edge analysis.
library(xml2) # this is for extraction of html table; results of leading edge analysis.
library(openxlsx) # for read xlsx file.
library(ComplexHeatmap)
library(tidyverse)
library(data.table)
library(survminer)
library(survival)
library(gridExtra)
library(MASS)
library(coin)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(RhpcBLASctl)
# # shiny
# library(shiny)
# library(bslib)
# Check global environment
Sys.getenv()