2025/12/14(日);初稿
2025/12/29(月);訂正;カプランマイヤー法による生存率の解析;教科書(生存期間解析、ISBN978-4-254-12861-1)に従って、打ち切りの設定を確認し、生存曲線(カプランマイヤー推定値)の横軸を1825日(5年)に限定した。
2026/01/03(土);訂正;元の記事のタイトル「TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.41, 2024)(正常組織のバイオリンプロットなし、GDC portalからのデータのダウンロード方法なし )」が長すぎてプラグイン「codoc」が正常に動作しなくなる可能性があったので、現在のタイトルに変更した。
2026/01/12(月);追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う。
2026/06/13(土);記事が長すぎてCodecプラグインが動かなかったので、コードをSTORESで販売することにした。
- 1 はじめに
- 2 解析の流れ
- 3 注意点1
- 4 注意点2
- 5 注意点3
- 6 ここからのRのダウンロード(文章を読みたくない人用)1
- 7 使用するパッケージ
- 8 RNA-seqのカウントデータを読み込む
- 9 ここからのRのダウンロード(文章を読みたくない人用)2
- 10 サンプル情報と診断情報を読み込む
- 11 カウントデータ、メタデータ、診断情報を関連付ける
- 12 データをAnndataで保存する(Optinal)
- 13 ここからのRのダウンロード(文章を読みたくない人用)3
- 14 解析のためのデータを読み込む
- 15 Geneset enrichment analysis (GSEA)に使用するGMTファイルを作成する
- 16 Geneset enrichment analysis (GSEA)に使用するGCTファイルを作成する
- 17 症例毎にGSEAを行ってエンリッチメントスコアを計算する
- 18 各種GSEAの結果をまとめる
- 19 疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する
- 20 疾患毎の遺伝子発現量やエンリッチメントスコアをバイオリンプロットで比較する
- 21 カプランマイヤー法による生存率の解析
- 22 データセット全体を保存する
- 23 追加の解析
- 24 まとめ
- 25 2026/01/12(月)追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う
はじめに
以前、TCGA(The Cancer Genome Atlas)に登録されたデータをダウンロードしたが、ここではTCGAのデータを使って各疾患毎の患者の生存率をカプランマイヤー法により解析する方法を述べる。これらはがんや腫瘍の遺伝子発現や遺伝子変異とそれに紐づく患者の情報であり、他の解析(基礎実験など)で疾患に関連しそうな遺伝子やタンパク質が見つかった場合、それらと疾患との関連性、特にそれらが患者の生存率に影響が及ぶかどうかを解析することが出来る。
解析の流れ
TCGAなどの解析で非常に面倒なのか、RNA-seqのカウント値が記載されたtsvファイル(のファイル名)と、それに対応する個々の患者の情報を結びつける作業が必要なことである。そのために、列名を適切に直し、不必要な重複を取り除かなければならない。このデータのクリーニングとも言える作業の後に、カウント値を種々の手法を使って解析し、最後に全解析結果と患者の情報を結合する。ここでは、そのカウント値を使ってGSEAなどを行い、遺伝子発現プロファイルの特徴を各患者(症例)毎に求め、それらを集計、必要ならば統計解析し、最後にそれらの特徴からある遺伝子や遺伝子セットに着目した生存率を求めて、関心のある遺伝子の発現量や遺伝子セットのエンリッチメントの程度が、患者の生存率にどれだけ関連するかを解析する。
注意点1
同じような記事(後日にポストした記事・新しい記事)を別に書いている。この記事との内容の違いは以下の表の通りである。もしこの記事を購入する場合は、以下の表を確認して、どちらが必要なのかを判断していただきたい。比較対象は新しい記事であるが、その解析に使ったTCGAのデータのバージョン(v.43)は、この記事のバージョン(v.41)よりも新しい。ただし、症例数はそんなに大きく変わらなかった。新しい記事にはssGSEA2とfgseaによるエンリッチメント解析が載っていない。この記事ではMsigDBのH: hallmark gene setsだけなのに対し、新しい記事ではMsigDBに登録のあるヒトの遺伝子セットを全部使ってエンリッチメント解析を行っている。また、新しい記事では正常組織のバイオリンプロットとの比較があったり、患者の生存期間に統計的有意な影響を与える遺伝子をまとめていたりするので、参考にはなると思う。
| 項目 | この記事 | 別の記事(後日にポスト) |
|---|---|---|
| タイトル | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.41, 2024) | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.43, 2025) |
| TCGAのバージョン | v.41, 2024 | v.43, 2025 |
| GDC portalからのデータのダウンロード方法 | なし | あり |
| GMTファイルの作り方 | あり(ただし、Geneontologyから選んできた遺伝子セット1つだけ) | なし |
| 使ったGMTファイル | Geneontologyから選んできて自分で作成したHRR(Homologous Recombination Repair)と、MsigDBのH: hallmark gene sets(Human MSigDB v2025.1.Hs、2025年10月時点)のみ。 | MsigDBに登録されているヒトの遺伝子セット全部(Human MSigDB v2025.1.Hs、2025年10月時点) |
| エンリッチメント解析 | ssGSEA2 escape GVSA fgsea | GSVA escape |
| バイオリンプロットによる疾患毎の遺伝子発現量もしくは遺伝子セットのエンリッチメントの比較 | 常組織なし ウィルコクソンの順位和検定なし | 正常組織あり ウィルコクソンの順位和検定あり(ただし、正常組織の例数が腫瘍組織に比べて少ないのでちょっと注意が必要と思う。) |
| 生存期間解析でのログランク検定 | あり | あり |
| その他 | 比較するデータの数(リストの要素の数)がそんなに多くないので、データの準備がそんなに大変じゃない。 | 比較するデータの数(リストの要素の数)が多く、そのために少しデータの準備がすこし複雑でメモリを喰いまくる。 |
注意点2
この解析では要求されるコンピューターの性能は案外高い。この解析で使用したCPU、RAM(Random Access Memory)(物理メモリ)のサイズ、SWAPのサイズは、それぞれRyzen 9 9950X、128GB 、2TBである。特に、RAMのサイズが大分と効く。もはや256GBあっても十分ではない。なので、SWAPを充分に用意しておかなくてはならない。SWAPについては、以前cellrangerを使っているときにSWAPファイルだとどうしてもメモリリークみたいなことになっていたので、2TBのm.2全部をSWAPパーティションとして与えている。こんな無駄に大きい理由は、本来別の用途でこの2TDのm.2を購入したのだが、結局本来の用途に不要になってしまい、それを流用したためであり、大きいからと行って無駄に1TBにSWAPパーティション、1TBにデータの保存とか設定するのが面倒だったからである。言うても、一般的には必要ではないのだが、逆にこれだけあればメモリについては不安になることもない(SWAP Thrashing;スワップスラッシング問題はある)。これが一杯になる計算があったとしたら、その計算では何かしらのエラー(例えば、スパースマトリックスとして扱うべきものを、普通のマトリックスとして計算してしまったとか、一つあたり100GB以上もあるデータセットに対する条件分岐をforeachで50000回も回してしまったなど。これについては何かしらの方法がありそうだけど、自分はそのあたり良くわからん。)が出ている可能性があるし、エラーではないとしたら自分が知っているものでは、シングルセルRNAシークエンスの解析においてtradeseq(あるクラスター間のトラジェクトリーに沿った遺伝子発現解析に使用するパッケージ)くらいである。データセットのサイズが大きいので、RAMの容量は多い方が良い、というか、少なすぎるとTCGAのような大きいデータセットの取り扱いは無謀と言える。以前も書いたが、これはRの弱点と思う。Rは計算しようとするデータセットを全部RAMに入れて計算しようとするので、例えば、Rではデータセットのサイズが500GBならば、RAMはシンプルに500GB以上必要になる。こういう場合、コンピューターに積んであるRAMの容量が16GBとかで、SWAP(Windowsでは「仮想メモリ」と呼ばれている)を500GBとか確保しないと、データがRAMに入り切らずに、入ったとしても計算出来ずにRもしくはコンピューターがクラッシュすると思う。また、こんなに大きなデータセットに対してGSEA(Gene Set Enrichment Analysis)等を行う場合は、ある程度性能の高いCPUも必要である。これがなければ、計算は出来るがそれに1ヶ月費やす、とかになる。
また、自分はこれらの解析をなんとCore i5、16GBのSurface Pro 8(5年以上前のコンピューター)でやっていたが、この手のCPUファンなどの冷却機能が劣るコンピューターを使うときは要注意である。Surfaceなどはファンはあるにしても、コンピューターの背面全体も使って熱を放出するような、なんとも受け身な冷却方法を採っており、これだと驚くほどコンピューターが熱くなる。こういったラップトップでここに書くような解析をしてしまうと、熱でラップトップの寿命を縮めるのは間違いない。
個人的にはメモリと冷却が十分であれば、5年以上前のコンピューターでも十分に解析に使用出来ると思っている。むしろ、CPU等は多少古いほうがパッケージとの相性などは良い可能性もある。もちろん、少し古いコンピューターではRAMが詰めなかったりするので、その場合はサーバーとかになるのだろう。所属する研究室や会社が許せば東大のSHIROKANEとか使ったほうがコストパフォーマンスが良いような気がする。その場合は自分で構築していいのか確認しなければならないような気がする。
また、ハードウェアで最近思うこととして、Rだけを使うならばIntelのCPUでWindowsを使うのが一番良いのではないかと思う。最近ではインテルへ不信感とCPUの性能の悪さからAMDが使用されがちであるが、これらのパッケージが開発・作成されたときに用いられていたCPUはインテルであり、インテルのCPUの方が相性が良いように思う。最近のMacは使ったことがないのでよくわからない。一方、自分はダウンロードやインストールが楽なのでUbuntuを使う。結局、なんやかやで痒いところに一番手が届くのがUbuntuである。
注意点3
この解析は、「疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する」の章までは本当に面白くない。それまではデータを読み込んだり、クリーニングしたり、整えて結合したりするだけである。途中のssGSEA2、escape、GSVA、fgseaを使ったGSEA(Gene Set Enrichment Analysis)があるが、整えたマトリックスなりGCTファイルを関数に入れるだけである。実際に解析やプレゼンテーションに使えそうな結果は、「疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する」以降からである。
以下、順次解析を始めていく。ちなみに、以下はすべてUbuntu上でのコードであるが、おそらくMacでもほぼ同じコードが動くはずである。Windowsの場合はパスの書き方が違うので注意が必要である。おそらく正規表現をエスケープさせる必要がある。なお、カプランマイヤー法云々はここでは述べないので、それらを知りたい場合は他のウェブページ、もしくはChatGPTにでも聞いたほうが色々と知ることが出来ると思う。
途中、Rのコードをダウンロード出来るようにしたので、読むのが面倒な場合はそのコードをダウンロードして、ディレクトリを適宜変えながら解析すれば良いと思う。また、インストールするパッケージ、使用しているコンピュータ、OSによってはエラーが出るときもあることを了承いただきたい。連絡をくれれば「出来ることであれば」サポートしようと思っている。
ここからのRのダウンロード(文章を読みたくない人用)1
あと、TCGAのカウントデータはここには含めていない。必要ならばTCGAから直接ダウンロードする必要がある。TCGAからカウントデータのダウンロードの方法はここに記載してある。
使用するパッケージ
以下のパッケージを読んでおけば、遺伝子発現量の解析であれば十分である。自分はこの環境で遺伝子発現の解析を行なっている。ここに、適宜必要なパッケージを追加しながら、解析を進めていく。
基本はtidyverseでデータを整えて、survivalでカプランマイヤー法を計算し、survminerで可視化する、という流れである。可視化にはgridExtraで各グラフをパネルにする。またdata.tableは作成したデータの読み込みに使用する。生存率の解析としては、基本的にこれくらいしか使わない。これらに加えて、fread、doParallelなどは読んでおいた方が良い。これらはfgseaのように、通常ならばシングルコアで計算する(実はBiocParalelが使用されている。全く調整できない、もしくは、自分にそんなスキルはないので、非常に邪魔くさい。)けど、だからこそ並列化した場合には効果が絶大と言える。もしシングルコアで計算しようものなら、一つのコードチャンクに1週間とか費やす羽目になってしまう。他のパッケージで重要な物は、GSEAやエンリッチメント解析に使用する物、ヒートマップ作成に使用する物などである。
また、この解析では最後にSys.getenv()で、Rの環境変数を出力している。理由はこの解析では途中にダウンロードして整形したTCGAの各患者の遺伝子発現プロファイルに対しssGSEA2、escape、fgsea、GSVAを適用するが、そのときに並列化がうまく行かず、そのトラブルシューティングで環境変数を確認したためだ。通常ならばこれは不要だが、別に最初に表示しない理由もないので今回は確認した。
# DEG
library(baySeq)
library(edgeR)
library(DESeq2)
library(limma)
# enrichment analysis
library(clusterProfiler)
library(fgsea)
library(ssGSEA2) # devtools::install_github("nicolerg/ssGSEA2") # devtools::install_github(broadinstitute/ssGSEA2.0) and devtools::install_github(broadinstitute/ssGSEA2) did not work.
library(escape)
library(GSVA)
library(pathview)
# immune cell deconvolution
library(ConsensusTME)
library(estimate)
library(immunedeconv)
library(MCPcounter)
library(msigdbr)
library(quantiseqr)
library(TIMER) #devtools::install_github('hanfeisun/TIMER'); it will not work!! use TIMER of immunodeconv if you need.
library(xCell)
# stats
library(NSM3) # sudo apt install libgmp-dev libgmp10 libgmp3-dev
library(pwr)
library(missForest)
# basic
library(beeswarm)
library(circlize)
library(org.Hs.eg.db)
library(org.Mm.eg.db)
library(doParallel)
library(foreach)
library(parallel) # Need it for use of baySeq
library(pheatmap)
library(rtracklayer)
library(rvest) # this is for extraction of html table; results of leading edge analysis.
library(xml2) # this is for extraction of html table; results of leading edge analysis.
library(openxlsx) # for read xlsx file.
library(ComplexHeatmap)
library(tidyverse)
library(data.table)
library(survminer)
library(survival)
library(gridExtra)
library(MASS)
library(coin)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(RhpcBLASctl)
# # shiny
# library(shiny)
# library(bslib)
# Check global environment
Sys.getenv()
RNA-seqのカウントデータを読み込む
まずはTCGAからダウンロードしたRNAシークエンスのカウントデータを全部読み込む必要がある。TCGAからのデータのダウンロードについては、この記事に詳しく書いた。データはそのときにダウンロードしたファイルである。以下の画像のように、大量のディレクトリに保存してあるTSVファイルを全部読み込む。それがRNA-seqのカウント値である。
このディレクトリに保存してあるファイルを全部読み込んで、all_fileに入れ、それを使ってファイル名の最後がcount.tsvで終わっているファイルのパスを全部取ってきてfile_pathに入れる。
# code 1
all_file <- list.files("/mnt/seqdata/public_data/Blog/TCGA/count", all.files = TRUE, full.names = TRUE, recursive = TRUE)
file_path <- all_file[str_detect(pattern = "counts.tsv$", string = all_file)] # 42348 rowsfile_pathに入っているパスから、str_detect()を使ってカウントデータ(TSVファイル)のファイル名を抜き出す。ファイル名は正規表現を使って抽出する。”^.*/”はドットなんとかというで終わっている部分という意味になる。ちなみに、自分が欲しい条件が書いてある正規表現を正しく記述するためには、ChatGPTに勝るものはないので、それを使ったほうが賢い。自分で考えて書いても良いのだが、考えるのに時間がかかるし、頑張って書いたとしてもChatGPTの方が圧倒的に正確である。ChatGPTに限らず、GeminiでもCopilotでも、自分の好きなサービスを使えば良いと思う。このファイル名が保存されたファイルは、後にカウントデータの列名をつけるために使用する。
# code 2
file_name <- sub("^.*/", "" , file_path) %>% data.frame()file_pathには、カウントデータまでのフルパスが入っているので、それを使ってカウントデータを取得していく。カウントデータは以下のようなフォーマットになっている。最初の6行までがライブラリ全体に関する情報が書いてある。
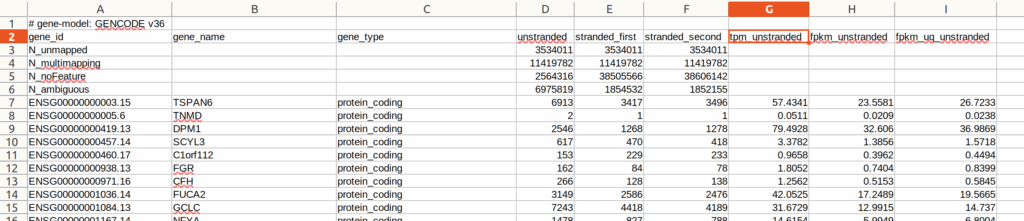
まず、読んでくるデータの入れ物、リストcountを定義する。この手のデータのハンドリングにはリストを使うのが一番良い。リストであれば、当然個々のデータにもアクセスしやすいし、それら全部に同様の処理をするのも容易である。自分はリストを使うためには、使う前にからのリストを必ず用意している(count<- list())。
カウントデータをread.table()でセパレーターをタブ(sep = “\t”)にして一気に読んで、リストcountに順番に入れていく。カウントデータの一行目には# gene-model: GENCODE v36とある。頭に#が付いてあるので、これはコメントアウトされる。したがってread.table()で読み込まれるのは2行目以降になり、そしてhead = TRUEがあるため、2行目のgene_idとある部分が列名として読み込まれる。つまり、read.table()がデータとして読んでくるのは3行目からになる。
ところが、3行目から6行目まではライブラリサイズなどが記載されている。自分で正規化したいひとはこれらが必要であるとは思う。この解析では既にTPM(transcript per million)で正規化された値であるtpm_unstrandedを使う。TPMは多施設から得られたデータの正規化には有用であると思う。そこで、元データの3行目から6行目まで、すなわち、読んできたデータの1行目から4行目までを除き(count[[i]] <- count[[i]][-c(1,2,3,4),])、実際の解析に必要な列gene_id、列gene_name、列tpm_unstrandedを選ぶ(count[[i]] <- count[[i]][,c(1, 2, 7)])。ただし、これらは最終的に一つに結合しなくては行けないので、区別をつけるために列tpm_unstrandedの部分をfile_nameを使ってファイル名にする。
最後reduce()で一気に結合し、それをwrite_tsv()で出力しておく。こんな時間が掛かる処理を毎回やっているのがダルいので、ファイルに出力した次第である。
ここで、Rに元から入っているwrite.table()とかと、tidyveseとかdplyrのwrite_tsv()とかの違いについて理解しておくのが良い。tidyverseの関数ってのは基本tibble型、百歩譲ってデータフレーム(こっちだとときどきエラーが出るので「百歩譲って」という表現を用いた。)しか扱えない。なので、ちょっと変なデータだったり、変な処理だったりする場合は。不便だが汎用性が高いwrite.table()を使うのが良いと個人的に考えている。経験的に。
こういった重い処理をやった後は、gc()を2回以上流しておまじないをしておく。
# code 3
count<- list()
for (i in 1:length(file_path)) {
count[[i]] <- read.table(
file = file_path[i],
sep = "\t",
header = TRUE)
count[[i]] <- count[[i]][-c(1,2,3,4),]
count[[i]] <- count[[i]][,c(1, 2, 7)]
colnames(count[[i]]) <- c("gene_id","gene_name", file_name[i,])
} # This takes so long time if you are using HDD.
count_merge <- purrr::reduce(count, inner_join)
write_tsv(count_merge, "/mnt/seqdata/public_data/Blog/TCGA/output/TCGA_primary_count_merge.tsv")
gc()
gc()
ここからのRのダウンロード(文章を読みたくない人用)2
サンプル情報と診断情報を読み込む
次に、Rのファイルを変えて、上記で保存したカウントデータを結合したTSVファイルを読み込む。このような大きなデータセットを読み込むためにはfread()が便利である。tidyverseとかdplyrのread_tsvとかよりもちょっとだけ早い。しかし、ここで注意点がある。fread()でTSVファイルをデータフレームとして読み込む場合、data.table = FALSEとする必要がある。デフォルトではTRUEであり、この場合はなんとテーブル(表)として読み込まれてしまう。テーブルとして読んでしまうと、inner_join()とかfull_join()が正常に動作しない。
# code 4
# Read each files. column name is for each case.
original_count <- fread(
input = "/mnt/seqdata/public_data/Blog/TCGA/output/TCGA_primary_count_merge.tsv",
sep = "\t",
encoding = "UTF-8",
data.table = FALSE)データフレームoriginal_countの3列目から最後の列までの列名をデータフレームにし、その列名をcase_idとして、データフレームcaseに入れる。この列名には、例えば”88215dd0-5841-44f1-9393-eefd8238cbb3.rna_seq.augmented_star_gene_counts.tsv”のような、カウント値を保存したファイル名が入っているはずである。
これは後ほど、他のメタデータを結合するためのメインキー(軸)になるので、重要なデータになる。よって、このデータフレームcaseにおける重複を確認しておく。ある変数の重複を除くには、データフレームの場合はdplyrのdistinct()が便利だが、これは重複を除くための関数(というか、Rの場合、関数というのだろうか….わからんけど、この記事ではこれらを関数と呼ぶ)であり、重複を確認するための関数ではない。重複を除くのではなく、重複があるかどうか確認するためには、確認のためのデータセットを新しく作った方が良いように思う。そのためには、データフレームcaseの列case_id(カウント値を入れたファイルのファイル名を入れた変数)をgroup_by()にいれて、グループ化して、それがそのグループが1つ以上あるcase_idをfilter()で拾ってきて、その行数をnrow()で示し、その値をcheck_case_duplicatedという値に入れれば良い。今回、このcheck_case_duplicatedの値には何もなかったので、ファイル名が重複していることはないと確認できた。これは当然である。もし重複があったら、ファイル名が同じものが別にダウンロードされてきたことになり、それは同じ患者の同じ部位からのシークエンスの結果が、なんらかの別のIDで重複して登録していることになる。それはなかなか起こり得ないし、ダウンロードの時点で起こっていたらきっと混乱してしまうだろうと思う。
# code 5
# Extract each case and check if dataset contains duplicated case.
case <- colnames(original_count)[3:ncol(original_count)] %>% data.frame() %>% rename("case_id" = ".")
check_case_Duplicated <- case %>% group_by(case_id) %>% filter(n() > 1) %>% nrow() # 0 case. There is no duplicated case
ここで、他の臨床情報、メタデータを読んでいく。色々と読んでいくが、空欄とかも随分と多く、生存率の解析で最低限必要な情報がそんなに多くはない。生存率の解析に必要な情報は解析や研究ごとに違うので、適宜必要な情報を調べて、それで抽出していく必要がある。
ここでの注意書きとしては、メタデータが2つのファイルに分かれている点についてである。TCGAは10000caseしかダウンロードできないので、このときは必要な症例数を2回にわけてダウンロードしたためである。ここで出来上がったファイルも、やはり出力しておく。ここではwrite_tsv()で良い。十分シンプルである。
# code 6
# Read all metadata
aliquot <- list()
analyte <- list()
clinical <- list()
exposure <- list()
family_history <- list()
follow_up <- list()
pathology_detail <- list()
portion <- list()
sample <- list()
slide <- list()
samplesheet <- list()
aliquot[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/aliquot.tsv")
aliquot[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/aliquot.tsv")
aliquot_merged <- purrr::reduce(aliquot, rbind)
analyte[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/analyte.tsv")
analyte[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/analyte.tsv")
analyte_merged <- purrr::reduce(analyte, rbind)
clinical[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/clinical.tsv")
clinical[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/clinical.tsv")
clinical_merged <- purrr::reduce(clinical, rbind)
exposure[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/exposure.tsv")
exposure[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/exposure.tsv")
exposure_merged <- purrr::reduce(exposure, rbind)
family_history[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/family_history.tsv")
family_history[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/family_history.tsv")
family_history_merged <- purrr::reduce(family_history, rbind)
follow_up[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/follow_up.tsv")
follow_up[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/follow_up.tsv")
follow_up_merged <- purrr::reduce(follow_up, rbind)
pathology_detail[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/pathology_detail.tsv")
pathology_detail[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/pathology_detail.tsv")
pathology_detail_merged <- purrr::reduce(pathology_detail, rbind)
portion[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/portion.tsv")
portion[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/portion.tsv")
portion_merged <- purrr::reduce(portion, rbind)
sample[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/sample.tsv")
sample[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/sample.tsv")
sample_merged <- purrr::reduce(sample, rbind)
slide[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/slide.tsv")
slide[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/slide.tsv")
slide_merged <- purrr::reduce(slide, rbind)
samplesheet[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/1/gdc_sample_sheet.2024-10-19.tsv")
samplesheet[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/2/gdc_sample_sheet.2024-10-19.tsv")
samplesheet_merged <- purrr::reduce(samplesheet, rbind)
カウントデータ、メタデータ、診断情報を関連付ける
TCGAでもCPTACでも何でも、カウントデータと個々のデータを何らかの値で結びつける必要がある。まず、samplesheetの列名に入っているスペースを除き、打つのが面倒なので全部小文字にしてしまう。
# code 7
# Change column name of samplesheet for later sataset preparation.
samplesheet_merged <- samplesheet_merged %>% rename(
"case_id" = "Case ID",
"sample_submitter_id" = "Sample ID",
"file_id" = "File ID",
"file_name" = "File Name",
"data_category" = "Data Category",
"data_type" = "Data Type",
"project_id"="Project ID",
"sample_type" = "Sample Type")clinicalには多くの欠損値があるが、それが「’–」という、一体どんな言語で使われているんだ?みたいな値が入っているので、それはRにおける欠損値NAに訂正しておく。
次に、カウント値(のファイル名)と患者情報を結びつけるために必要な値、さらに、生存率の解析に必要そうな値のみを選んで新しくデータフレームclinical_merged_required_survivalを作成する。ここでやったことは、欲しいカラム名を保持したデータフレームrequired_values_for_surivivalを作成し、それを使ってdplyrのselect()で必要な列を抽出してくる。
# code 8
# Clean up clinical.tsv
# Replace "'--" to "NA".
clinical_merged[clinical_merged == "'--"] <- NA
colnames(clinical_merged)
# Clinical information must be include survival period. Therefore, the dataset must be filtered by that survival data is true.
required_values_for_surivival <- c(
"case_id",
"case_submitter_id",
"project_id",
"days_to_death",
"days_to_last_follow_up",
"age_at_diagnosis",
"days_to_birth",
"vital_status",
"year_of_birth",
"year_of_death",
"year_of_diagnosis",
"days_to_treatment_end",
"days_to_treatment_start",
"treatment_outcome",
"treatment_type",
"primary_diagnosis",
"site_of_resection_or_biopsy",
"tissue_or_organ_of_origin",
"diagnosis_is_primary_disease",
"days_to_last_known_disease_status",
"days_to_recurrence",
"classification_of_tumor")
clinical_merged_required_survival <- clinical_merged %>% select(all_of(required_values_for_surivival))そして、列days_to_deathがNAではない、または、列days_to_last_follow_upがNAではない患者情報を抜き出してきて、データフレームclinical_merged_required_survival_2を作成する。これらはつまり、フォローアップもされていないか、死亡日の記録がないか、なので、生存率の計算には使えなさそうだ。
この解析では患者が死んでいないか(列days_to_death)、最後のフォローアップの日(列days_to_last_follow_up)を使ってカプランマイヤー法で生存率を解析するが、このあたりを目的に応じて変える必要がある。そのためにも、一体どんな変数が何かをわかっておく必要がある。
# code 9
clinical_merged_required_survival_2 <- clinical_merged_required_survival %>%
filter(days_to_death != "NA" | days_to_last_follow_up != "NA")生存率の解析に使えそうな患者を抜き出してきてデータフレームclinical_merged_required_survival_2を作成したわけだが、ここでも重複した情報がないか確認する。重複が合った場合、それはどんな情報なのかを確認しなくてはならないので、dplyr::distinct()ではなくgroup_by() >%> filter(n()>1)を使う。dplyr::distinct()を使うとデータフレームから自動で重複がなくなってしまい、どれが除かれたのか、そもそも除いても良かったのかが確認出来ないためである。
そうすると、19954例もある。
個々の患者の識別用の値が入っているのは複数あるので、次に同様のことをcase_submitter_idを使ってやってみると、やっぱり19954例もあった。なんてこった。
重複している症例を見てみると、どうやら同じ患者由来の検体のようだ。違いは、列treatment_typeに入っている値が”Pharmaceutical Therapy, NOS”だったり”Radiation Therapy, NOS”だったりである。つまり、これらの重複例は異なる治療を受けている同じの患者のようだ。check_case_idを見てみると、列days_to_deathや列days_to_last_follow_upには同じ値が入っていた。色々と考えたが、生存率の解析で一番重要なのはフォローアップの期間(列days_to_last_follow_up)が計算されていること、活きているか死んでいるか(列days_to_death)の情報である。これらの値が同じであれば、これらの重複はシンプルに除去しても問題はなさそうだ。
# code 10
# "case_id" holds the data like "004d251-3f70-4395-b175-c94c2f5b1b81".
# extract the record with duplicated "case_id."
check_case_id <- clinical_merged_required_survival_2 %>% group_by(case_id) %>% filter(n() > 1)
nrow(check_case_id) # 19954 cases
# "case_submitter_id" holds data like "TCGA-DD-AAVP".
# extract the record with duplicated "case_submitter_id".
check_case_submitter_id <- clinical_merged_required_survival_2 %>% group_by(case_submitter_id) %>% filter(n() > 1)
nrow(check_case_submitter_id) # 19954 casesよって、ここで一気にdplyr::distinct()を使って、これらの重複(異なる治療法が別々に登録されている同一の患者)している一方を除くことにする。実際にdplyr::distinct()を解析に使用するデータセットに当てる前に、重複が解消されているかどうか確認するためのデータセットを作り、それを使って、2つの別のID(列case_id、列case_submitter_id、別のファイル由来の値)それそれで重複例を除き、同じだけ症例数が残るかどうかを確認する。何かあったときのために、.keep_all = TRUEにして、変に列が削除されないようにする。
# code 11
check_case_id_distinct <- clinical_merged_required_survival_2 %>% distinct(case_id, .keep_all = TRUE)
nrow(check_case_id_distinct) # 9926 rows
check_case_submitter_id_distinct <- clinical_merged_required_survival_2 %>% distinct(case_submitter_id, .keep_all = TRUE)
nrow(check_case_submitter_id_distinct) # 9926 rows症例数が同じだったので、ようやくdplyr::disitct()で重複している一方を除き、新しくデータフレームclinical_merged_required_survival_de_duplicatedを作成する。ここでも.keep_all = TUREを入れて、重複の判定に使った変数(列名)が無駄に除去されてしまわないようにする。ここで作るデータフレームclinical_merged_required_survival_de_duplicatedが、カプランマイヤー法による生存率の計算に使用する打ち切りの値を設定するために必要なデータセットとなる。
# code 12
clinical_merged_required_survival_de_duplicated <- clinical_merged_required_survival_2 %>% distinct(case_id, .keep_all = TRUE)
nrow(clinical_merged_required_survival_de_duplicated) # 9926 rowsここは本解析にはあまり関係ないが、どんな疾患がどれだけあるのかを確認しようと思う。そのためには、集計したい列名をtable()に入れ、次に、その出力をdata.frame()に渡して、それをオブジェクトにしておいたら良い。ここでは、列classification_of_tumor、列primary_diagnosis、列tissue_or_organ_of_originについて集計をとって、それぞれdf_から始まるデータフレームに入れておいた。そのうち、言うてこの時点では使うかどうかわからないのだが、データフレームdf_primary_diagnosisが後の解析で有用に見えたので、それをwrite_tsv()で保存しておいた。
# code 13
# Check what kind of tumor type is there.
table(clinical_merged_required_survival_de_duplicated$classification_of_tumor)
# I think all values of "classification_of_tumor" is about primary tumor. For example, if the value is "metastasis", it means that metastatic tumor was found as "primary tumor". Therefore, there is no need to filter the data with classification_of_tumor.
# These data.frames are for check what kind of cancer type is registered??
df_primary_diagnosis <- table(clinical_merged_required_survival_de_duplicated$primary_diagnosis) %>% data.frame()
df_tissue_or_organ_of_origin <- table(clinical_merged_required_survival_de_duplicated$tissue_or_organ_of_origin) %>% data.frame()
df_diagnosis_x_tissue_or_organ_of_origin <- table(clinical_merged_required_survival_de_duplicated$primary_diagnosis, clinical_merged_required_survival_de_duplicated$tissue_or_organ_of_origin) %>% data.frame()
write_tsv(df_primary_diagnosis, "/mnt/seqdata/public_data/Blog/TCGA/output/df_diagnosis.tsv")続いて、使ったサンプルに関する情報を整理する。データフレームsample_mergeを作ったが、それを使う。このデータフレームsample_mergeには、使ったサンプルの由来の臓器、腫瘍なのか正常組織なのか(今回は残念ながら正常組織のデータをダウンロードしていない。)、固形臓器なのか血液なのか等の情報が保持されている。これを、上記で作成した、生存期間などが書かれたデータフレームclinical_merged_required_survival_de_duplicatedと結合すべく、上記と同じように重複を除く作業を行う。
まず、欠損値が「’–」という謎の値として入っているため、それをNAに置き換える。次にtable()を使ってどの列にどんな値が入っているか確認する。その結果、どうやら列tissue_typeと列sample_typeにより、生存率の解析に有用なデータだろうということがわかった。列tissue_typeにはそのサンプルが腫瘍組織由来なのか(tissue_type=Tumor)、正常組織由来なのか(tissue_type=Normal)が入っおり、sample_typeには、それが原発巣由来(sample_type=Primary Tumor)なのか、血液由来(sample_type=Blood Derived Cancer – Peripheral Blood)なのか、転移巣由来(sample_type=Metastatic)なのか、その他の原発巣由来なのか(sample_type=Additional – New Primary)などの値が入っている。この解析では、filter()を使って列tissue_typeがTumorのサンプルを拾ってきてデータフレームsample_merged_2を作成し、次に列sample_typeがPrimary Blood Derived Cancer – Peripheral BloodもしくはPrimary Tumorのサンプルを拾ってきてデータフレームsample_merged_3を作成する。
ここでちょっと余談だが、自分は色々と確認しながらデータフレームを作成していくが、すでにどの値を使って何を選んでくるかが明らかであれば、sample_merged_2やsample_merged_3のようにいちいち新しいデータフレームを作成しながら解析を進める必要はない。全部sample_mergeに上書きしても一向にかまわん。しかし、こうやってステップバイステップで拾っていけば、何かやってしまったときでもそこに戻れるので多少は便利と思う。ただし、RAMを着実に消費していくので、このように大きなデータセットの場合は要注意である。最終的にRAMが圧迫され、そしてSWAPを使い出し、最終的にSWAP Thrashingが発生して計算がめちゃくちゃ遅くなることは間違いない。
必要なサンプルを抽出したら、次に上記の解析でもあったように、重複例がどれだけあるか、どのような重複が生じているか確認する必要がある。ざっと見た感じではsample_submitter_idがサンプルのIDとして特有そうなので、それを使ってgroup_by()でグループ化し、n()により1つ以上のカウントの値を重複例として抽出し、確認用のデータフレームとしてchecK_sample_submitter_idを作成する。確認したところ、どうやらこの重複は、組織を採取した時期や採取方法(FFPEかどうか)などに違いがあるようだ。今回の解析ではそのあたりはどうでも良い(いや、本当はどうでも良くない。解析の目的に合わせて、一番最初に採取した時期のデータ、病理のレポートがあるサンプル、FFPEじゃなくてOCTでのサンプルと一致させたりする必要がある)ので、気にしないで解析を進めることにした。最終的に各データフレームはproject_id、case_submitter_id、sample_submitter_id、case_idで結合していく。
よって、この解析では、シンプルにdistingt()でsample_submitter_id(これにはTCGA-BK-A0CCなどの値が入っている)の値の重複を解消し、新しくsample_merge_4というデータフレームを作成する。今考えれば、列case_idでも良かったように思う。
# code 14
# Clean up sample
# Replace "'--" to "NA".
sample_merged[sample_merged == "'--"] <- NA
# Check number of case
nrow(sample_merged) # 30343
# If peripheral blood, bone marrow, and tissue were collected from a patient, the dataset "sample_merge" contains 3 records for each individual.
# Check what kind of speciemens are in dataset "sample_merged".
table(sample_merged$sample_type)
# Check what kind of specimens are in dataset "sample_merged". At first, looking at "tissue_type" suggest that there are information of tumor and normal tissue (probably) surrounding tumor.
table(sample_merged$tissue_type)
# There are 11060 normal tissues, and 19283 tumor tissues.
# In this analysis, we are interested in tumor tissue. Therefore, normal tissue will be removed.
# the metadata contains clinical (individual) information, but the RNA-seq count was not downloaded before. therefore normal will be removed.
sample_merged_2 <- sample_merged %>% filter(tissue_type == "Tumor")
# The output suggested that several metastatic, FFPE, recurrent, and new primary tumor were included in the dataset. Our interest is "primary tumor" and therefore "Primary Blood Derived Cancer - Peripheral Blood" and "Primary Tumor" will be left.
table(sample_merged_2$sample_type)
sample_merged_3 <- sample_merged_2 %>% filter(sample_type == "Primary Blood Derived Cancer - Peripheral Blood" | sample_type == "Primary Tumor")
# Check if column "sample_submitter_id" in dataset "sample_merged_3" contains all unique or duplicated values.
checK_sample_submitter_id <- sample_merged_3 %>% group_by(sample_submitter_id) %>% filter(n() > 1)
# There are 302 duplicated cases.
# In this analysis, purpose of dataset "sample_merge" is for the matching each case and count data. Therefore, I think de-duplication is good enough for further analysis. If you need, for example, pathological report (which is stored at column "pathlogy_report_uuid") or, you need to analyze the difference between sample collection days (which is stored at column "days_to_collection"), you need to get back here to check which cases are required for the analysis.
# Therefore, duplicated cases will be simply removed from the dataset "sample_merged_3".
# I think we need to use the information that are associated with RNA-seq count, which is linked with "file_name".
sample_merged_4 <- sample_merged_3 %>% distinct(sample_submitter_id, .keep_all = TRUE)ここで、これまで作成したデータセットの行数と列数、つまり、症例数と臨床情報の項目数を確認した。ここまでいろんな値を見てきて、既にどのデータフレームにどんな値を入れているか忘れてしまっている。特に症例数を確認する必要があるが、各データフレームで値が違っていても構わないはずである。だって、各データフレームでは解析対象として抽出してくるときの基準が異なる。例えば、患者の生存期間を保持しているデータフレームclinical_merged_required_survival_de_duplicatedではフォローアップされていて生存期間が記載されている症例を選んでいるわけだし、データフレームsample_merged_4では、同一患者になるようにフィルターされている。ここでは、nrow(samplesheet_merged)(すなわち、カウント値のファイル数、列gene_name;遺伝子名と列gene_id;ensemblの遺伝子IDの2列が追加されていることに注意)とncol(original_count)(症例数)が一致していれば良い。むしろ、この2つが一致していない場合、それは、TCGAからのダウンロードが自体が何かおかしいってことになる。
# code 15
# Required dataset for further analysis.
# count dataset
original_count # this is original count data, which compose fine_name as column.
ncol(original_count) #10081 (10079 cases + gene_id + gene_name)
# samplesheet_merged
samplesheet_merged
nrow(samplesheet_merged) # 10079 cases
# clinical_merged_required_survival_de_duplicated
clinical_merged_required_survival_de_duplicated
nrow(clinical_merged_required_survival_de_duplicated) # 9926 cases
# sample_merged_4
sample_merged_4
nrow(sample_merged_4) # 19014 records, because there are several different information for a case. ここから、上記でクリーニングしてきた各データフレームを結合すべく作業をしていく。データフレームsamplesheet_mergeには、カウント値のファイル名と列case_id、列case_submitter_idなど、他のデータフレームと結合するときのメインキー(Rでこのような言い方をするかどうかはわからん。SASではメインキーと言っていたはず。)が含まれている。後にカウント値(行もしくは列名がカウント値のファイル名になるはず)を結合するために必要なデータフレームと言える。まず、データフレームsamplesheet_mergedの後々にメインキーとなり得る列でソートしていく。ソートにはarrange()を使う。同様に、上記で作成したデータフレームsample_merged_4もメインキーになりそうな列でソートしておく。
話は外れてしまうが、ここでarrange()によるソートが必要だった理由を述べる。当初、データセットをAnndataオブジェクトとして後の解析を行なうつもりだったためである。しかし、結局RでPytyonのパッケージを使うのは効率的ではないことがわかり、結局このソートはデータセットのQC的な目的しか果たさなくなってしまった….ちょっと面倒な手順が加わってしまったと思っている。
話を作業に戻す。次に、データフレームsamplesheet_mergedとデータフレームsample_merged_4を列sample_submitter_idをメインキーにしてinner_join()で結合し、新しくデータフレームsample_samplesheetを作る。ここで、両データフレームには同じような列名が入っているため、それを区別するためにsuffix = c(“_sample”, “_samplesheet”)を入れて、どの列がどちらのデータフレーム由来だったのか区別出来るようにする。
nrow(sample_samplesheet)で列数(すなわち症例数)を確認すると、10079例ある。
データフレームsamplesheet_mergedとデータフレームsample_merged_4の両方を列project_id、列case_id、列sample_submitter_idでソート(arrange())したので、これを使って結合前のデータフレームと一緒の順番になっているか、確認する。当然、なっているはずであるが、このあたりはやっておいて損はないと思う。それが# check order以下である。やることとしては、データフレームsamplesheet_merged(カウント値のファイル名が入っているデータフレーム)の列sample_submitter_id(これはソート済)をベクトルorder_sample_submitter_idとして、データフレームsample_samplesheet(さっき作成したサンプルの情報が入っているデータフレーム)の列sample_submitter_id(これもソート済)をベクトルcheck_sample_submitter_idとして保存する。その2つのベクトルをデータフレームcheck_sample_orderにする。そしてそれら2つのベクトルは同じように並んでいればOK、そうでなければNGを入れていく。最後に、table()で全部OKであることを確認するという具合である。
次に、データフレームsample_samplesheetにあるsample_submitter_id列がどれだけ重複しているかどうか確認してみると、なんと79例も重複しているじゃあないか。それに、どうもこれらはカウント値のファイル名も違っている…なんか、これはやばそう….と思ったけど、この作業の最後このデータフレームsample_samplesheetを後々、患者の生存期間が保持されたデータフレームclinical_merged_required_survival_de_duplicatedとinner_join()するが、そのときに、生存率が記録されている症例と結合するので、おそらく大丈夫なはず。というか、この2つの重複を区別するための列がfile_idであるが、この値を、患者のサンプル等の情報が保持された各データフレーム(aliquot、analyte、clinical、exposure、family_history、follow_up、pathology_detail、portion、sample、slide、samplesheet)で検索しても、それを保持してる列を見つけることができなかった。だから、ちょっと見切り発車感はあるが、データフレームclinical_merged_required_survival_de_duplicated(すでに全部ユニーク、つまりこれらは同一の患者から得られていることが確認できている)とinner_join()してできたデータフレームを、同一の患者の原発巣から得られたもっともらしいカウント値とそれに紐付けられた患者情報を保持したデータフレームとして解析を進めていくしかなさそうでだった。一方、データフレームsample_samplesheetにある列file_nameには、重複した値はなかった。
# code 16
# At first, merge "samplesheet_merged" and "sample_merged_4" by column "sample_submitter_id".
# order the files
# each value looks like;
# samplesheet_merged$project_id; TCGA-ESCA
# samplesheet_merged$case_id; TCGA-IG-A97I
# samplesheet_merged$sample_submitter_id; TCGA-IG-A97I-01A
samplesheet_merged <- samplesheet_merged %>% arrange(project_id, case_id, sample_submitter_id)
# Match column "sample_submitter_id" in dtaset "sample_merged_4" and column "sample_submitter_id" in dataset "samplesheet_merged".
# sample_merged_4$project_id; TCGA-LUSC
# sample_merged_4$case_submitter_id; TCGA-NC-A5HJ
# sample_merged_4$sample_submitter_id; TCGA-NC-A5HJ-01Z
sample_merged_4 <- sample_merged_4 %>% arrange(project_id, case_submitter_id, sample_submitter_id)
# Merged samplesheet_merged and sample_merged_4 by column "sample_submitter_id".
sample_samplesheet <- inner_join(sample_merged_4, samplesheet_merged, by = c("sample_submitter_id"), suffix = c("_sample", "_samplesheet"))
# then order the dataset.
sample_samplesheet <- sample_samplesheet %>% arrange(project_id_samplesheet, case_id_samplesheet, sample_submitter_id)
nrow(sample_samplesheet) # 10079 cases
# check order
order_sample_submitter_id <- samplesheet_merged$sample_submitter_id
check_sample_submitter_id <- sample_samplesheet$sample_submitter_id
check_sample_order <- data.frame(samplesheet = order_sample_submitter_id, sample_samplesheet = check_sample_submitter_id)
check_sample_order <- check_sample_order %>% mutate(
check =case_when(
samplesheet == sample_samplesheet ~ "OK",
TRUE ~ "NG"
)
)
table(check_sample_order$check) # 10079 cases are OK.
# check column "case_id_samplesheet"
check_unique_case <- sample_samplesheet %>% group_by(sample_submitter_id) %>% filter(n() > 1)
# there are different file name in a same case.
check_unique_file <- sample_samplesheet %>% group_by(file_name) %>% filter(n() > 1) # all unique filesカウント値のファイル名とそれに対応する患者の情報を入れたデータフレームsample_samplesheetと、患者の生存期間を入れたデータフレームclinical_merged_required_survival_de_duplicatedをinner_join()する。このとき、データフレームsample_samplesheetの列case_id_sampleと、データフレームclinical_merged_required_survival_de_duplicatedの列case_idをメインキーとして結合する。また、同じ列があった場合は、データフレームsample_samplesheet由来ならば、新しく出来る列の末尾に何も付けず(””)、データフレームclinical_merged_required_survival_de_duplicated由来ならば”_clinical”をつけることにする。次に、これまでと同様に、arrange()により行を重要と考えられる列でソートし、column_to_rownames(“file_name”)で列名をfile_nameにし、出来上がったデータフレームをarrange()により念の為もう一度重要と考えられる列でソートする。ここで出来上がったデータフレームsample_samplesheet_clinicalが、カウント値のファイル名と解析に必要な患者情報を保持したものであり、後の解析で使用するデータフレームである。
# code 17
# merge sample_samplesheet and clinical_merged_required_survival_de_duplicated
sample_samplesheet_clinical <- inner_join(
sample_samplesheet,
clinical_merged_required_survival_de_duplicated,
by = c("case_id_sample" = "case_id"),
suffix = c("", "_clinical")
)
sample_samplesheet_clinical <- sample_samplesheet_clinical %>% arrange(project_id_samplesheet, case_id_samplesheet, sample_submitter_id)
sample_samplesheet_clinical <- sample_samplesheet_clinical %>% column_to_rownames("file_name")
sample_samplesheet_clinical <- sample_samplesheet_clinical %>% arrange(project_id_samplesheet, case_id_samplesheet, sample_submitter_id)
患者情報のデータセットsample_samplesheet_clinicalが出来上がったので、次は遺伝子発現プロファイルのデータフレームを整えることにする。別に、データフレームoriginal_countをそのまま使ってもいいんだけど、やっぱり順番通りに並んだデータセットが欲しい。上述でデータフレームsample_samplesheetを重要と考えられる列でソートしたが、その順番で並んでいるファイル名を取ってくれば良い。このためにデータフレームsample_samplesheetの列file_nameをベクトルorder_file_nameとして作成し、そのベクトルをそのままselect()で使うことで、並び替えたデータフレームを取ってくることが出来る。このデータフレームをcount_reorderとした。このときに、後々に列名となり得る列gene_idと列gene_name(特に、列gene_name)も拾ってくることを忘れないようにする。そして、いちいち取ってきた症例数をlength()、nrow()、ncol()で確認したほうが、間違いにすぐに気づくことが出来て良い。
このあたりから、オブジェクトの数が多くなってくるので、一連の作業が終わったタイミングでgc()を入れたほうが良いように思う。gc()が2回あるのは、上述の通りおまじないである。
# code 18
# make sure order the dataset "original_count" based on the order of "file_name" in dataset "sample_samplesheet".
# this is for the ordering dataset "count" because the count matrix must be stored into anndata object and therefore order of file must be consistent with all dataset.
order_file_name <- sample_samplesheet$file_name
length(order_file_name) # 10079 cases
# reorder the dataset "original_count" based on the order of "file_name" in dataset "sample_samplesheet".
count_reorder <- original_count %>% select(c("gene_id", "gene_name", all_of(order_file_name)))
ncol(count_reorder) #10081 (10079 cases + gene_id + gene_name)
gc()
gc()データフレームcount_reorderは各症例における遺伝子発現量がカウント値として保持されている。これはTPM(Transcripts per million)で正規化されたカウント値なので、各症例間の比較が可能である。ここでもやはり、重複した遺伝子があるかどうかを確認する。そうすると110個の遺伝子に重複があった。それらを何とかしてデータフレームcount_reorderから除く必要がある。
そのために、まず、重複した遺伝子を拾ってくる。それには、table()で列gene_nameを集計し、
それをデータフレームに入れ、カウントが1つ以上の遺伝子をfilter()で拾って来れば良い。後々、データフレーム(厳密に言えばtibble型でdplyrを使いたい。基本的にdplyrはデータフレームに使えるが。ときどきデータフレームに対してdplyrの関数を使うとエラーが出るが、これはtibble型じゃない、とかの理由のときがある。)としてdf_gene_nameとして取っておく。そして、このデータフレームdf_gene_nameを使って、データフレームcount_reorderから重複のある遺伝子のみで構成されるデータフレームcout_reorder_QCを作る。
データフレームcount_reorder_QCを眺めていると、どうやらY染色体にある遺伝子が無駄に重複しているようだ。それに、これらのカウント値は非常に低い、というか、ゼロっぽい。ということは、そもそも一般的には発現していない遺伝子なのかもしれない。なので、先ずはこのY染色体に由来する遺伝子を除き、次に、低いカウント値の遺伝子を除くことにした。先ず、Y染色体に由来する遺伝子を除くことにする。str_detect()を使って列gene_idに入っている値(これはensemblの遺伝子ID。遺伝子名が重複していても、ensenblの遺伝子IDは固有のはず。)の末尾に「_PAR_Y」が付いているものを除く。実際には、「_PAR_Y」が付いている遺伝子「以外」を拾ってくるようにコードを書いた。これによりデータフレームcount_reorder_QC_1を作成した。次に、カウント値が全症例に渡ってゼロの遺伝子を除くことにする。このためにはおなじみrowSums()で全症例で合計が0の遺伝子を除けば良い。こうして作成したデータフレームをcount_reorder_QC_2とした。しかしながら、まだ一部の遺伝子の重複が残っているようだ。そこで、それらのうち最大のカウント値の遺伝子、つまり、一般的にはなかなか発現してこないが、その中でもようやくカウントされてくるような遺伝子を残すことで、重複を解消した。これをデータフレームcount_reorder_QC_3とした。group_by(gene_name) %>% filter(n() > 1)で確認したところ、重複は解消されているようだ。
データフレームcount_reorder_QC_3には、元のcount_reorderにおいて重複している遺伝子のうち、解析に有用そうな遺伝子だけが入っている。データフレームcount_reorder_QC_3の列gene_idを使って、元のデータフレームcount_reorederからその遺伝子だけを拾ってきてデータフレームcount_reorder_QC4を、さらに、元のデータフレームcount_reorderで重複している遺伝子を全部除いてしまったデータフレームcount_reorder_no_duplを作り、この2つをrbind()で縦に結合することでデータフレームcount_reorder_post_QCを作った。これが、これで、重複が解消されたはずである。次にこのデータフレームでもgroup_by(gene_name) %>% filter(n() > 1)で重複した遺伝子名が実際に無いことを確認し、ensenblの遺伝子IDを除いてデータフレームoriginal_count_mat_tを作成した。今後、これを使ってGSEAなどを計算していく。
それ以降は、遺伝子名の重複を解消したデータセットcount_reorder_post_QCは、確かに遺伝子の重複がないか、カウント値がゼロのみの遺伝子が含まれていないか、などを確かめるための作業であり、各データフレーム(count_reorder_post_QC_2、count_reorder_post_QC_4、count_reorder_post_QC_4)である。
# code 19
# check are duplicated gene names in dataset "count_reorder".
# There are a lot of duplicated genes (110 genes).
df_gene_name <- table(count_reorder$gene_name) %>% data.frame() %>% rename("gene_name" = "Var1") %>% filter(Freq > 1)
nrow(df_gene_name) # 110 duplicated genes
# check_gene_name <- count_reorder %>% group_by(gene_name) %>% filter(n() > 1)
# select the duplicated genes
count_reorder_QC <- count_reorder[count_reorder$gene_name %in% df_gene_name$gene_name == TRUE,]
# remove genes at chromosome Y
# "PAR_Y" means the PseudoAutosomal Regions (PAR) of chromosome Y.
# https://www.gencodegenes.org/pages/faq.html
count_reorder_QC_1 <- count_reorder_QC %>% filter(str_detect(gene_id, "_PAR_Y") == FALSE)
# calculate sum of count and percent of 0 over the cases for every genes.
count_reorder_QC_1 <- count_reorder_QC_1 %>% mutate(
sum_count = rowSums(count_reorder_QC_1[,3:ncol(count_reorder_QC_1)]),
percent_zero_count = apply(count_reorder_QC_1[,3:ncol(count_reorder_QC_1)], 1, function(x) (sum(x == 0)/length(x)))
)
# # sum(x == 0) returns the number of 0 in a row.
# # following is practice to count number of 0 in a row.
# test_df <- data.frame(a = c(0,0,0,1,1), b = c(0,0,1,1,1), c = c(1,0,0,0,1))
# sum(test_df[1,] == 0)
# sum(test_df[4,] == 0)
# Of the genes in dataset "count_reorder_QC_1", remove no gene count in any cases.
count_reorder_QC_2 <- count_reorder_QC_1 %>% filter(sum_count != 0)
# Of the genes in dataset "count_reorder_QC_2", leave genes with maximum rowSum value in each gene.
count_reorder_QC_3 <- count_reorder_QC_2 %>%
group_by(gene_name) %>%
filter(sum_count == max(sum_count))
# check if the genes in dataset "count_reorder_QC_3" are duplicated or not.
check_duplicated_genes <- count_reorder_QC_3 %>% group_by(gene_name) %>% filter(n() > 1)
nrow(check_duplicated_genes) # 0
# count_reorder_QC_1, _2, and _3 contain "sum_count" and "percent_zero_count" columns. These columns are not required for the analysis.
count_reorder_QC_4 <- count_reorder[count_reorder$gene_id %in% count_reorder_QC_3$gene_id == TRUE,]
# remove duplicated genes from dataset "count_reorder".
count_reorder_no_dupl <- count_reorder[count_reorder$gene_name %in% count_reorder_QC$gene_name == FALSE,]
check_duplicated_genes_most_part <- count_reorder_no_dupl %>% group_by(gene_name) %>% filter(n() > 1)
nrow(check_duplicated_genes_most_part) # 0
# merged all unique genes.
count_reorder_post_QC <- rbind(count_reorder_no_dupl, count_reorder_QC_4)
check_duplicated_genes_post_QC <- count_reorder_post_QC %>% group_by(gene_name) %>% filter(n() > 1) # There is no duplication.
nrow(check_duplicated_genes_post_QC) # 0
# remove gene_id
count_reorder_post_QC_2 <- count_reorder_post_QC %>% select(-one_of("gene_id"))
# store order of gene_name
order_gene_name <- count_reorder_post_QC_2$gene_name
# This is final count matrix
original_count_mat <- count_reorder_post_QC_2 %>% column_to_rownames("gene_name")
original_count_mat <- original_count_mat %>% select(all_of(rownames(sample_samplesheet_clinical)))
original_count_mat_t <- t(original_count_mat)
# check which genes are significantly low counts over all cases.
## This is used for anndata.
count_reorder_post_QC_3 <- data.frame(gene_name = count_reorder_post_QC_2$gene_name)
count_reorder_post_QC_3$rowSums <- rowSums(count_reorder_post_QC_2[,2:ncol(count_reorder_post_QC_2)])
count_reorder_post_QC_3$percent_zero <- apply(count_reorder_post_QC_2[,2:ncol(count_reorder_post_QC_2)], 1, function(x) (sum(x == 0)/length(x)))
count_reorder_post_QC_3 <- count_reorder_post_QC_3 %>% column_to_rownames("gene_name")
## This is used for dataframe.
## If some calculation for each genes, use this dataframe.
count_reorder_post_QC_4 <- count_reorder_post_QC_2 %>% column_to_rownames("gene_name")
count_reorder_post_QC_4$rowSums <- rowSums(count_reorder_post_QC_4[,2:ncol(count_reorder_post_QC_4)])
count_reorder_post_QC_4$percent_zero <- apply(count_reorder_post_QC_4[,2:ncol(count_reorder_post_QC_4)], 1, function(x) (sum(x == 0)/length(x)))これでカウント値(のファイル名)と患者情報が対応出来たので、次にそれらを結合する。まず、view(head())、nrow()、ncol()で各データフレームがどういった感じなのか、それらの列数、行数を確認する。ここで、以降はもしかしたらhead()を使った後にview()をやった方が良いと思う。特にこのように大きいマトリックスをview()だけで表示しようとすると、大体めちゃくちゃに遅い。それにRstudioがクラッシュすることがかなり多い(自分は)。データフレームは一応大丈夫だが、それでも遅い場合が多い。なので必要以上に大量のデータを表示する必要はない。
上記で遺伝子発現プロファイルをデータフレームoriginal_count_mat_tを作り、rowname_to_columns()でその行名(カウント値のファイル名)を列file_nameに移動させる。同じように、患者情報を保持したデータフレームsample_samplesheet_clinicalも行名を列file_nameに直す。そして、作成したこの2つのデータフレームoriginal_count_mat_t_dfとsample_samplesheet_clinical_dfを、メインキーを列file_nameにしてinner_join()にし、データフレームorigianl_df_w_infoを作る。このデータフレームorigianl_df_w_infoが、実際に解析に用いるデータフレームである。
# code 20
view(head(original_count_mat_t)) # matrix
view(head(sample_samplesheet_clinical)) # obs
view(head(count_reorder_post_QC_3)) # var
# Matrix
nrow(original_count_mat_t) # 10017 cases
ncol(original_count_mat_t) # 59422 genes
# Observations
nrow(sample_samplesheet_clinical) # 10017 cases
ncol(sample_samplesheet_clinical) # 66 conditions
# Variables
nrow(count_reorder_post_QC_3) # 59422 genes
ncol(count_reorder_post_QC_3) # 2 variables
# matrix and observations can be simply combined.
## at first, original_count_mat_t needs to be converted into data frame
view(head(original_count_mat_t))
original_count_mat_t_df <- data.frame(original_count_mat_t)
view(head(original_count_mat_t_df))
nrow(original_count_mat_t_df) # 10017 cases
ncol(original_count_mat_t_df) # 59422 genes
## then combine original_count_mat_t_df and sample_samplesheet_clinical
original_count_mat_t_df <- original_count_mat_t_df %>% rownames_to_column("file_name")
sample_samplesheet_clinical_df <- sample_samplesheet_clinical %>% rownames_to_column("file_name")
origianl_df_w_info <- inner_join(
original_count_mat_t_df,
sample_samplesheet_clinical_df,
by = "file_name"
) %>% column_to_rownames("file_name")
nrow(origianl_df_w_info) # 10017 cases
ncol(origianl_df_w_info) # 59488 genes (59422 genes + 66 conditions)解析に使用するデータフレームorigianl_df_w_infoが整ったところで、今後グラフを見やすくするために疾患名を示す列を付け足すことにする。まず、TCGA-BRCAやTCGA-LUADなどの疾患名が入っている列project_id_samplesheetから、頭の「TCGA-」を削除して、BRCAやLUADという値が入っている列diseaseを作りたい。そのためにはgsub()を使って頭の「TCGA-」を””(何も入れない)に置き換えればよい。
次に、疾患毎に通し番号を入れたい。このために、直前で作成した列diseaseをgroup_by()でグループ化し、row_number()でそのグループ毎、つまり疾患毎に番号を振り、その番号をmutate()で新しい列individual_orderとして作った。次にその疾患毎の番号である列individual_orderと疾患名の列diseaseをunite()で結合して列disease_individual_orderを作った。
# code 21
origianl_df_w_info <- origianl_df_w_info %>% mutate(
disease = gsub("^TCGA-*", "", origianl_df_w_info$project_id_samplesheet),
order = c(1:nrow(origianl_df_w_info)))
origianl_df_w_info <- origianl_df_w_info %>%
group_by(disease) %>%
mutate(individual_order = row_number(disease))
origianl_df_w_info <- origianl_df_w_info %>% unite(col = "disease_individual_order", c("disease", "individual_order"), sep ="", na.rm = FALSE, remove = FALSE)
やっと解析を行うためのデータセットが出来た。生存率などの解析は、別のRマークダウンで行なっていくことにする。なので、出来上がったデータセットをTSVファイルで保存し、別のRマークダウンで改めて読み込んで使用することにする。そうしなければ、おそらくスワップスラッシングにより計算が一生進まなくなる(進むけどおそすぎてCPUが明らかに無駄になる)。
やることとしては、write_tsv()で保存すれば良いのだが、これは列名が保存されないので、rownames_to_column()でデータフレームの行名を列にしておく。
# code 22
# these are for survival analysis
origianl_df_w_info_write <- origianl_df_w_info %>% rownames_to_column("file_name")
count_reorder_post_QC_4_write <- count_reorder_post_QC_4 %>% rownames_to_column("gene_name")
write_tsv(origianl_df_w_info_write, file = "/mnt/seqdata/public_data/Blog/TCGA/output/origianl_df_w_info.tsv")
write_tsv(count_reorder_post_QC_4_write, file = "/mnt/seqdata/public_data/Blog/TCGA/output/count_reorder_post_QC_4.tsv")データをAnndataで保存する(Optinal)
ここは飛ばしても構わない。せっかくコードを書いたので、ここにも載せておく。
途中、arrange()を使って、使用するつもりのデータフレームを同じ順番にソートしたが、実はこの解析をやっているとき、「そう言えば、RでもAnndataがあったな…それ使ったらデータのハンドリングが大分と楽なんじゃあないのか…?」と思い、作成したデータフレームをAnndataオブジェクトで保存しようとか考えていたためである。本来入っているデータが一致しているならば、ソートする必要はない。そういう理由から、以下はAnndataで保存するための方法である。注意点としては、これまでの作業であった通り、カウント値をデータフレームではなくマトリックスで保存しなければならないこと、行名と列名をそれに合わせる必要があるため、データの順番は揃えた方が良さそうなことと、NAをnanに変えないとAnndataオブジェクトに出来ないことである。だから途中で、データフレームの名前がoriginal_count_matだったり、いちいちarrange()でソートして順番を合わせたり、という作業が入ったわけである。
しかしながら…
やってみてわかったのだが、RでAnndataを使うのは、非常に無駄だった。その理由は、結局Anndataで解析を行なっていくとなると。ReticulateでPythonのコーディングをする必要があった。それなら、最初っからPythonで行くわ。
# code 23
# dataset for obs, which means observation.
# "sample_samplesheet_clinical" will be "obs".
# rownames of "sample_samplesheet_clinical" is "file_name"
order_obs <- rownames(sample_samplesheet_clinical) %>% data.frame() %>% rename("file_name" = ".")
# dataset for var, which means variable.
# "count_reorder_post_QC_3" will be "var"
# rownames of "count_reorder_post_QC_3" is "gene_name"
count_reorder_post_QC_3
order_var <- rownames(count_reorder_post_QC_3) %>% data.frame() %>% rename("gene_name" = ".")
# check order of variables in "original_count_mat"
# "original_count_mat" will be X.
var_name_of_X <- rownames(original_count_mat) %>% data.frame() %>% rename("gene_name" = ".")
obs_name_of_X <- colnames(original_count_mat) %>% data.frame() %>% rename("file_name" = ".")
#
check_var <- cbind(var_name_of_X, order_var)
colnames(check_var) <- c("var_name_of_X", "order_var")
check_var <- check_var %>% mutate(
check = case_when(var_name_of_X == order_var ~ "OK",
TRUE ~ "NG")
)
table(check_var$check) # 59422 cases are OK.
#
check_obs <- cbind(obs_name_of_X, order_obs)
colnames(check_obs) <- c("obs_name_of_X", "order_obs")
check_obs <- check_obs %>% mutate(
check = case_when(obs_name_of_X == order_obs ~ "OK",
TRUE ~ "NG")
)
table(check_obs$check) # 10017 cases are OK.
# order of obs (observations) and var (variables) looks good. Anndata object will be prepared at next step.
# code 24
# Anndata is basically python library, and they can not handle "NA". Therefore, "NA" must be replaced with "nan".
sample_samplesheet_clinical[is.na(sample_samplesheet_clinical)] <- "nan"
# code 25
# sample_samplesheet <- sample_samplesheet %>% column_to_rownames("file_name")
# "original_count_mat_t" was prepared at line ~598.
library(anndata)
original_anndata <- AnnData(
X = original_count_mat_t,
obs = sample_samplesheet_clinical,
var = count_reorder_post_QC_3
)
# write_h5ad(original_anndata, "/mnt/seqdata/public_data/Blog/TCGA/output/original_anndata.h5ad")
ここからのRのダウンロード(文章を読みたくない人用)3
以降のコードは以下のリンクで販売している。ZIPを展開するとRのコードが3つあるので、そのうちの「3 2024 11 02 TCGA survival.Rmd」というファイルがここからのコードである。
解析のためのデータを読み込む
やっと解析に取り掛かる。遺伝子発現量だけで生存率を解析しても良いのだが、せっかく多くの患者の遺伝子発現プロファイルがあるので、いくつかシグナル経路のエンリッチメント解析を行い、その結果も使って生存率を解析する。
まずはデータを読み込まなくてはならない。そのためには、上述でもあったfread()を使ってデータフレームoriginalを作る。このとき、。data.table = FALSEとする必要がある。これをやらないとデータフレームではなく表として読み込まれてしまい、後々データの結合などが出来なくなってしまう。
データフレームoriginalは、上記で遺伝子発現プロファイルであるデータフレームoriginal_count_mat_t_dfと患者の臨床情報のデータフレームsample_samplesheet_clinical_dfを列file_nameでinner_join()したデータフレームである。これには各症例に通し番号を入れた列file_nameが付いている。その列file_nameは、ただの番号(1から10017)なので、パッと見てそれが患者の番号であることがわかるような文字列を付けたい。なので、caseという値のみが入っている列caseをmutate()で作り、次にunite()を使って各症例の通し番号のみが入った列file_nameと、作成した列caseを結合し、新しく列caseを作成した。
# code 26
# load data
load("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/2024 11 02 TCGA survival.RData")
# code 27
original <- fread(
input = "/mnt/seqdata/public_data/Blog/TCGA/output/origianl_df_w_info.tsv",
sep = "\t",
encoding = "UTF-8",
data.table = FALSE) # This data must be used for the analysis
original <- original %>% mutate(case = "case")
original <- original %>% unite(col = "case", c("case", "file_name"), sep = "", remove = TRUE)
# IMPORTANT NOTICE column "case" will be used for a main key for inner_join() after several enrichment analysis (ssGSEA2, escape, gsva, fgsea)Geneset enrichment analysis (GSEA)に使用するGMTファイルを作成する
ここで、なんの遺伝子セット(Geneset)についてGSEAしようかと考えた。とりあえず一つ考えたのが、Homologous Recombination Repair(相同組み換え修復)に関連する遺伝子でも集めるか、ということである。対象ががんなんだから、DNA修復には何かしら違いがあるだろって感じである。あまり悩まずに即席でやりたかったので、GOに行って、その遺伝子セットを集めてくる。まず、ここに行く(https://geneontology.org/)。そして、homologous recombination repairと入力して、関連するパスウェイを検索してみる。
そうすると、どうやら384個くらいありそう。そこで、右側の「Term」というところの一番上にあるdouble-strand bread repair via homologous recombinationをクリックした。
そうすると以下の画面になった。どうやら、おそらく9083個の遺伝子があるらしい。
次に、OrganismでHomo Sapiensのところの「+」を押す。
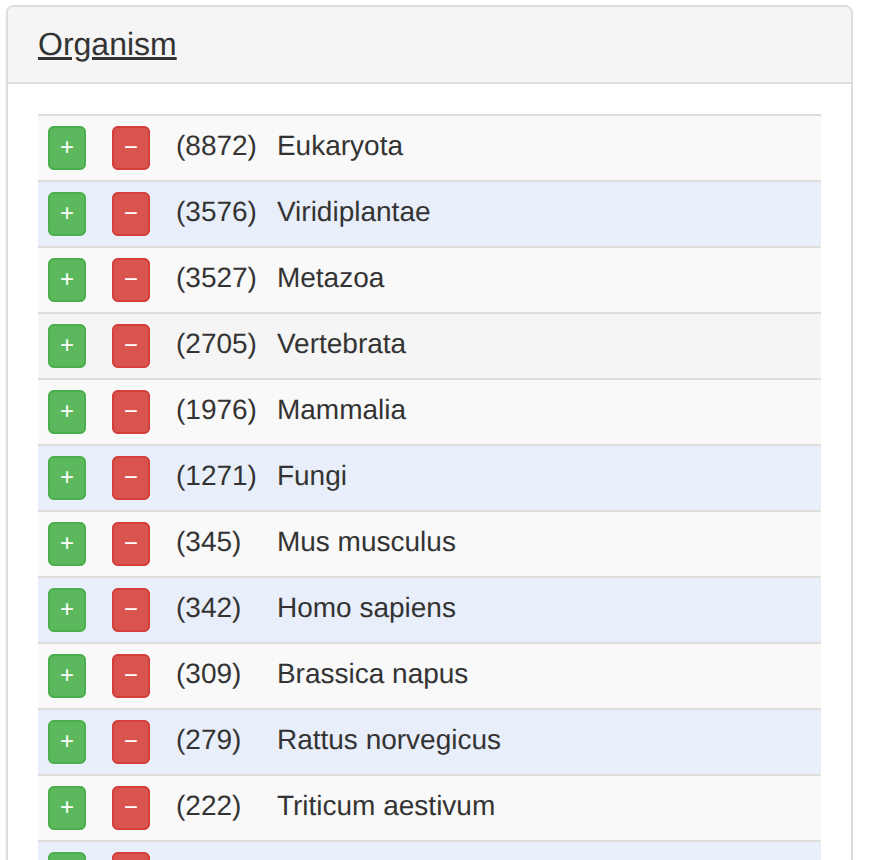
また、TypeのProteinのところの「+」も押す。
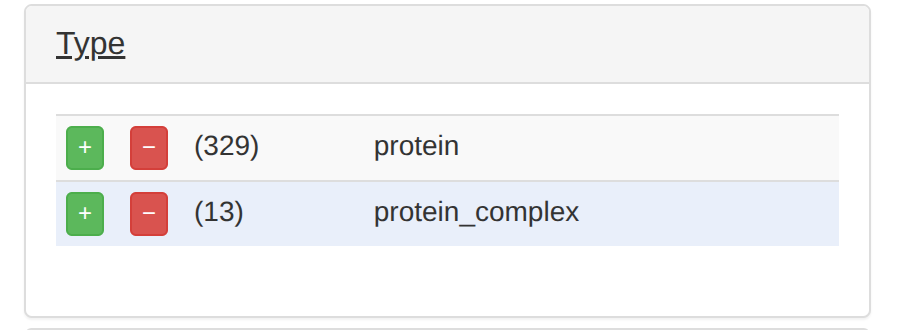
そうすると、遺伝子が329個まで絞り込めた。どうやらGO:0000724というパスウェイらしい。
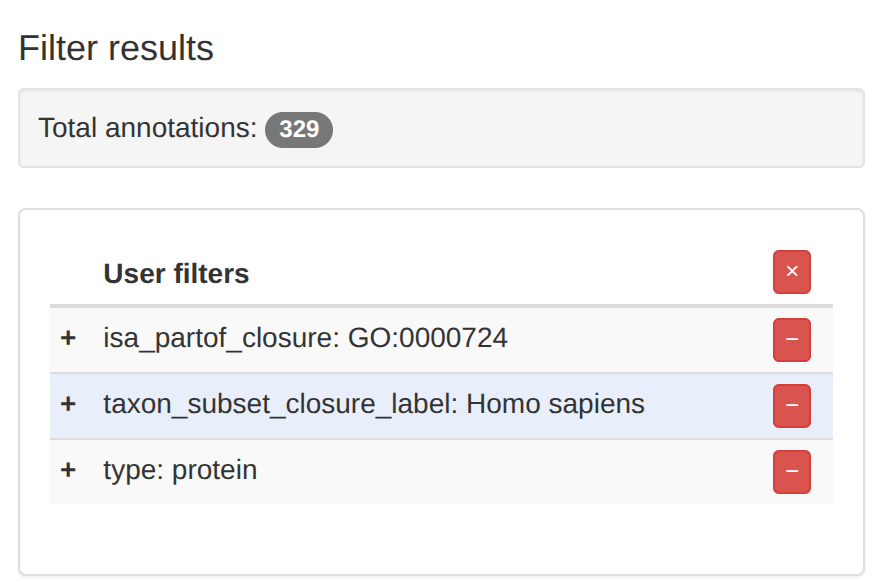
以下がGO:0000724に属する329個の遺伝子である。「Download」と書かれているボタンを押す。
そうすると、どんな情報をダウンロードするか選ぶことが出来る。この解析では、遺伝子名が必要なので、Available poolからGene/product(bioentity)を選んできて、それを表示する。
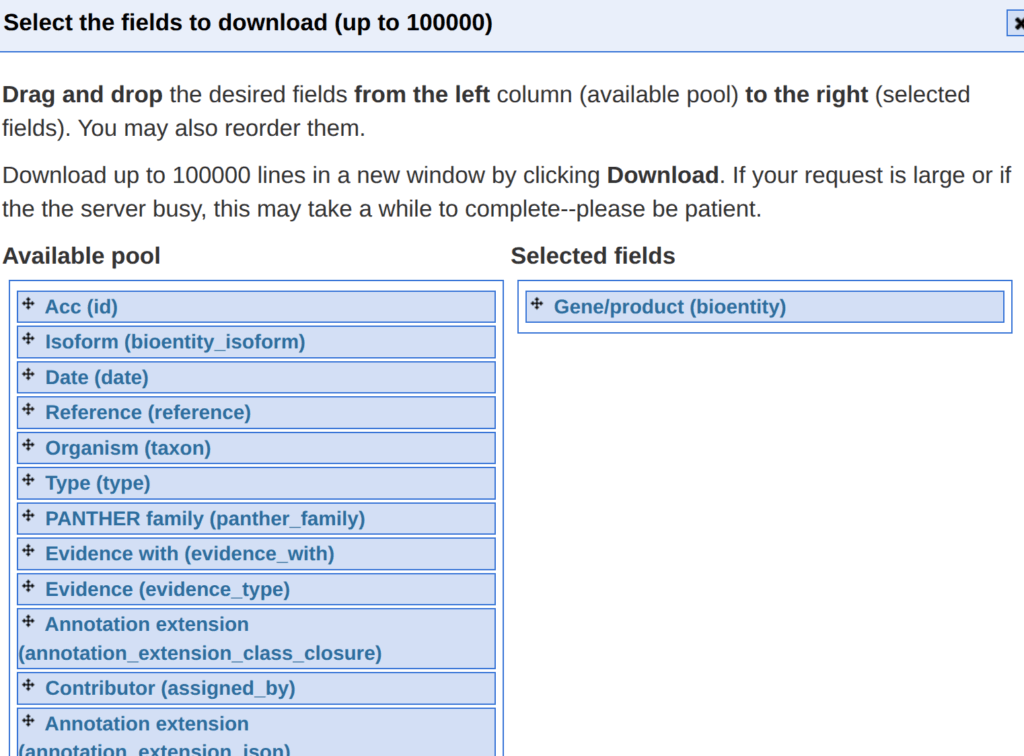
非常にイケてないのが、実質はダウンロード出来ない。たしか、ブラウザに表示されるだけである。これをコピーして、なんでもいいのでテキストファイル(.txt)として保存する。個々ではgeditに貼り付けてテキストファイルで保存した。というか、なんだこのUniprotKBは。遺伝子名にしろよって感じである。調べたが遺伝子名ではダウンロードできない。上述の表では遺伝子名だったのに。なんだこの使いにくいデータベースは。XXだな。
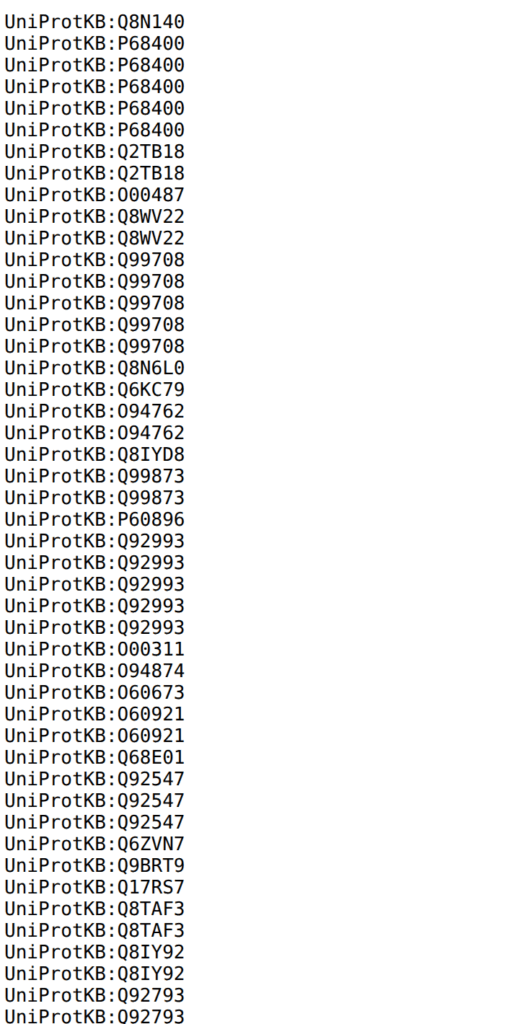
言うてても仕方ないので、RでuniprotのIDを遺伝子名に変換するとしよう。そのためには、UniprotKB:以降のIDをダブルクオーテーションで囲む必要がある。そのためにはgeditでCTRL+Hを押し、「UniProtKB: 」を「”」に置き換える。続いて、正規表現の改行「/n」を「”,」に置き換える。そうすると、UniprotKBのIDで書かれたベクトルが出来上がる。これをそのままRに貼り付ける。
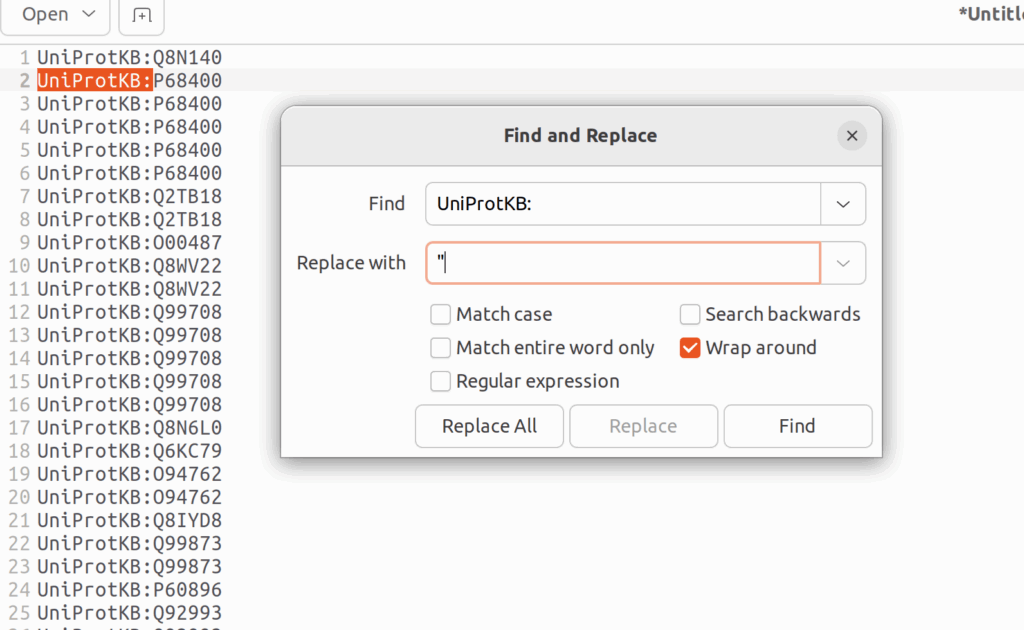
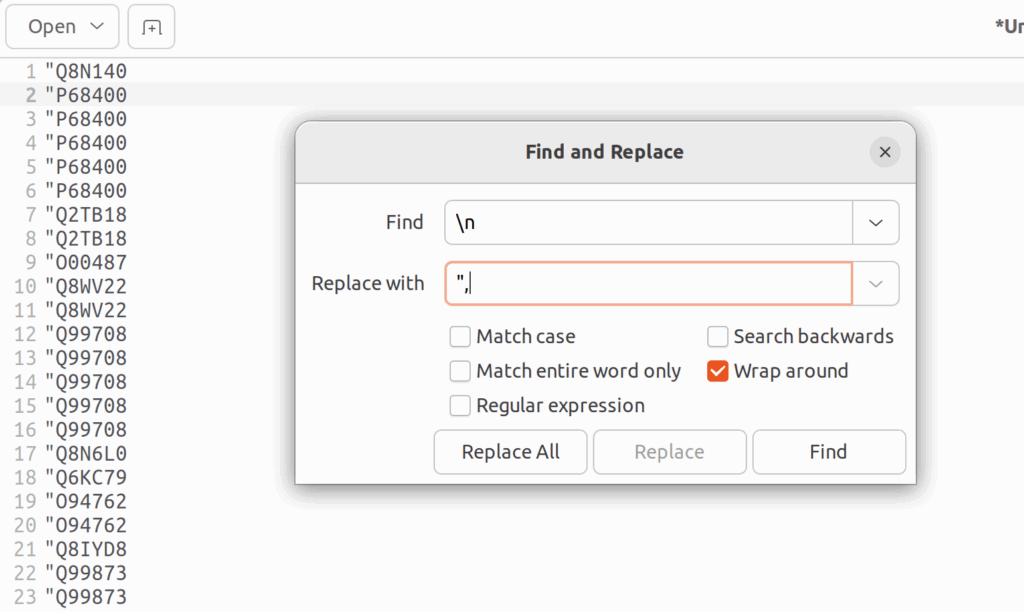
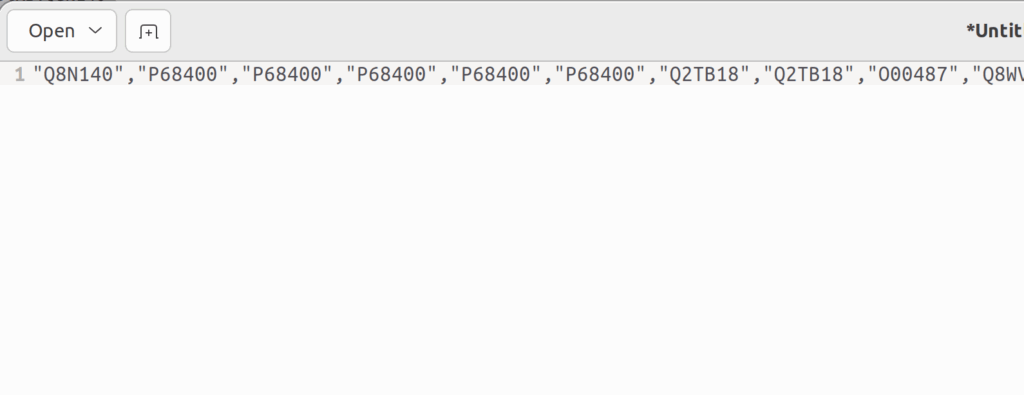
そして、AnnotationDbiというパッケージのmapIds()でUniprotIDを遺伝子名に置き換え、それをベクトルgeneset_HRRとして保存する。
# code 28
uniprot_id <- c("Q8N140","P68400","P68400","P68400","P68400","P68400","Q2TB18","Q2TB18","O00487","Q8WV22","Q8WV22","Q99708","Q99708","Q99708","Q99708","Q99708","Q8N6L0","Q6KC79","O94762","O94762","Q8IYD8","Q99873","Q99873","P60896","Q92993","Q92993","Q92993","Q92993","Q92993","O00311","O94874","O60673","O60921","O60921","Q68E01","Q92547","Q92547","Q92547","Q6ZVN7","Q9BRT9","Q17RS7","Q8TAF3","Q8TAF3","Q8IY92","Q8IY92","Q92793","Q92793","Q8IY18","Q8IY18","Q8IY18","Q9BV68","Q9BV68","Q8TDG4","Q8TDG4","Q92698","Q92698","Q8IX21","P33993","P33992","P33991","O60216","Q9BQI6","P39748","Q9BQ83","Q9BQ83","P49959","P49959","P49959","P49959","P49959","P49959","P49959","P49959","P49959","P49959","P49959","P49959","P49959","Q9BQ15","Q9BQ15","Q9BQ15","Q9BQ15","P49916","Q8N635","Q8N635","P23246","P53350","P53350","P53350","P53350","P53350","Q8WVD3","Q8WVD3","Q8WVD3","P43351","P43351","P43351","P43351","Q13315","Q13315","Q13315","Q13315","Q13315","Q13315","Q13315","Q96T88","P61088","Q9NVH0","Q13627","Q13472","Q9NXX6","Q96SB8","Q96SB8","Q9NXL9","Q9NXL9","Q9NXL9","Q9NXL9","Q9NXL9","Q9NXL9","Q13156","P46063","Q6ZRQ5","Q6ZRQ5","Q6ZRQ5","Q6ZRQ5","Q6ZRQ5","Q6ZRQ5","Q6ZRQ5","O60934","O60934","O60934","O60934","O60934","O60934","O60934","O60934","O60934","O60934","O60934","O60934","Q14159","Q14159","Q14159","Q14159","O75925","Q96NY9","Q9H7T9","O75771","O75771","Q9ULU4","Q9ULG1","Q9NRZ9","Q9NRY2","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q06609","Q9NUS5","Q9NUS5","Q9H9A7","Q9H9A7","Q07021","Q07021","O15315","O15315","O15315","Q96E14","Q9Y3Z3","Q9Y3Z3","Q9Y2M0","Q9Y2M0","Q8NCN4","Q9Y620","Q9Y620","Q9Y620","Q9Y620","Q8NFZ0","Q8NFZ0","Q9Y294","Q9Y294","Q9Y294","Q9UBU8","P27694","P27694","P27694","P27694","P27694","P27694","P27694","P27694","Q96B01","Q96B01","Q96B01","Q8NB91","P49736","Q96FA3","Q96HA7","Q96HA7","Q96HA7","Q96HA7","Q96HA7","Q96HA7","Q96HA7","Q9H1E3","Q9H1E3","Q9UJA3","Q9UJA3","Q9UJA3","Q9UJA3","Q9UJA3","O75419","Q9Y248","Q9UGP5","P54132","P54132","P54132","P54132","Q9H2F5","Q9H2F5","Q19AV6","Q19AV6","Q9H4I0","Q86YC2","Q86YC2","Q86YC2","Q86YC2","Q86YC2","Q86YC2","Q4G0Z9","Q4G0Z9","Q4G0Z9","Q9NQR1","P60510","O95072","Q8NAG6","Q8NHY5","Q05BQ5","Q05BQ5","P35244","P35244","P35244","Q96MG7","Q96MF7","Q96MF7","Q96MF7","Q96MF7","Q6PCD5","Q6PCD5","Q6PCD5","Q6PCD5","P25205","Q6B0I6","P09874","P09874","P09874","Q7Z3K3","P16104","P16104","O94761","P38398","P38398","P38398","P25490","Q14191","Q68DK2","Q68DK2","P28715","O43719","O43719","O43719","O43543","O43543","O43543","O43543","O43542","O43542","O43502","O43502","Q1ZZU3","Q1ZZU3","Q1ZZU3","Q7Z5Q5","Q14839","Q14566","Q14565","Q14565","Q96AH0","Q96AH0","Q96AH0","Q96AH0","O43299","P15927","P15927","P15927","Q92878","Q92878","Q92889","Q92889","Q99684","Q99684","P51587","P51587","P51587","P51587","P51587","P51587","Q86XK3","Q86XK3","Q86XK3","P78527","P78527","Q6NVH7","Q6NVH7")
geneset_HRR <- mapIds(org.Hs.eg.db,
keys = uniprot_id,
keytype = "UNIPROT",
column = "SYMBOL",
multiVals = "first")
geneset_HRR <- unique(geneset_HRR)
geneset_HRR変換した遺伝子名を使ってGMTファイルを作成する。cat()を使って、保存したベクトルgeneset_HRRを遺伝子セットの名前をHRR、コメントとしてGOのアクセッションIDであるGO:0000724、セパレーターとしてタブ(\t)、ファイル名としてgeneset_HRR.gmtとして保存する。これでオリジナルのGMTファイル(言うてパスウェイはHRRの一つだけであるが)を作ることが出来る。
それに加えて、MsigDB(https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp#H)からダウンロードしてきた遺伝子セットも読み込む。MsigDBに登録されているヒトの遺伝子セット(特定のシグナル経路や分子パスウェイのことをここでは遺伝子セットと呼ぶ)全部を、GSVAパッケージの関数であるreadGMT()でmsigdb_2025_1オブジェクトとして読み込む。しかしながら、この遺伝子セットのGMTファイルはかなり多いので、この解析では使用しない。じゃあ読むんじゃあねえよって感じしかしない。やれば出来るよってことで読み込んだ。いや、本当はそれを使って各種GSEAを使いたかったんだけど、多すぎるかなって感じがしたため、断念した。元気があるひとは、以下に述べるhallmark_2025_1ではなくmsigdb_2025_1を使ってみたら良い。一応ここで断っておく。おそらく、msigdb_2025_1を使った場合、計算時間が無限に思えるほど長いのでやめたほうが良いと思う。
そういうことなので、この解析では50個の遺伝子セットからなるH: human hallmark gene setsを使用する。これはhallmark_2025_1として読み込んだ。これを使用することにした理由は、MsigDBに登録されたヒトの遺伝子セット(H、C1からC8まで)のうち、遺伝子セットの数が少ないためであり、かつ、代表的なシグナル経路と考えられるためだ。
# code 29
cat("HRR", "GO:0000724", geneset_HRR, sep = "\t", file = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/geneset_HRR.gmt")
msigdb_2025_1 <- readGMT("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/msigdb_v2025.1.Hs_files_to_download_locally/msigdb_v2025.1.Hs_GMTs/msigdb.v2025.1.Hs.symbols.gmt")
hallmark_2025_1 <- readGMT("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/h.all.v2025.1.Hs.symbols.gmt")Geneset enrichment analysis (GSEA)に使用するGCTファイルを作成する
GSEAにはGCTファイルという各疾患の遺伝子発現量(TPM正規化されたカウント値)は書かれたファイルが必要になる。GCTファイルは以下の画像のようなフォーマットになる。ssGSEA2に入れるGCTファイルは、ここ(https://docs.gsea-msigdb.org/#GSEA/Data_Formats/)に記載されているフォーマットと少し違うので注意する必要がある。ssGSEA2に使用するGCTファイルは2列目にDescriptionが無い。もし2列目にDescriptionを入れてしまうと、ssGSEA2でエラーが出てしまう。
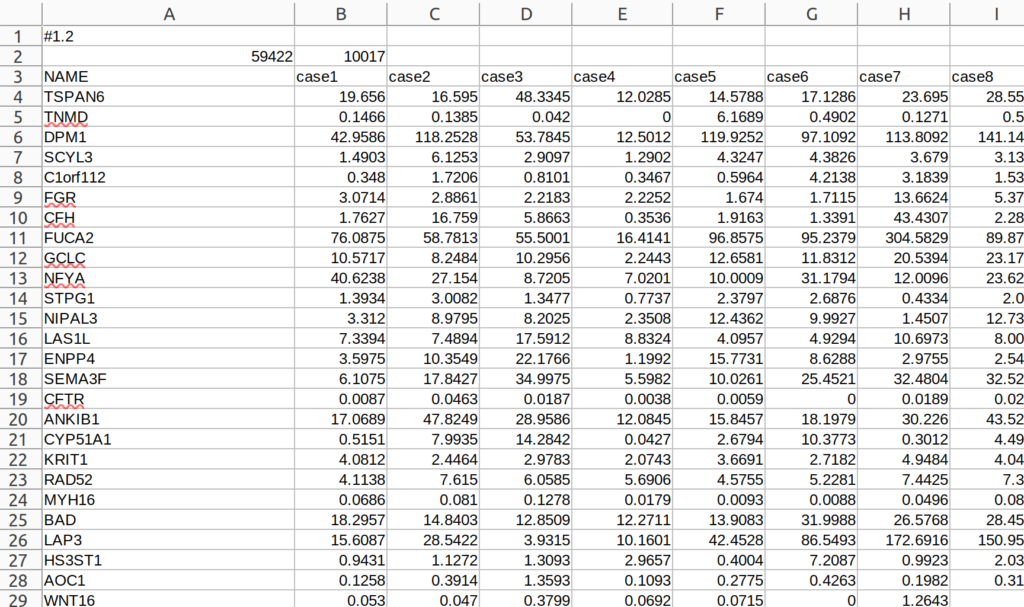
このGCTファイルの作成には、上記で作成した(というか、読み込んできた)データフレームoriginalを使用する。originalには、各症例の通し番号が振られている列caseがあるので、その列名をrename()でNAMEに直す。そして、その列NAMEをcolumn_to_rownames()でデータフレームの列名にする。それをt()で転置、すなわち、行列を入れ替える。データフレームに対してt()を使うと、出来上がってきたオブジェクトはマトリックスになってしまうので、data.frame()によりデータフレームに直し、それをデータフレームenrich_3_tとする。このデータフレームenrich_3_tの列名(各症例の通し番号)を改めて列NAMEとして戻しておく。これをデータフレームenrich_3_t_1とする。このデータフレームenrich_3_t_1は後ほどGCTファイルを作成するために使用する。
ここで、なぜこんなに列NAMEをデータフレームの列名にしたり、またデータとしての列に戻したりしているかというと、データフレームの行列をt()で入れ替えるときに文字を扱えなかったり、それをそのままデータフレームに戻すとその列名が消えてしまったりするためである。これはデータを変えるたびに一々確認しながら作業を進めるのが良いと思う。
GCTファイルは主にssGSEA2に使用するが、escape、GVSA、fgseaは通常のマトリックスで良い。なので、マトリックスenrich_3_t_mat(これがescape、GVSA、fgseaに必要)とenrich_3_mat(もしかしたら他のパッケージで使うかもしれないので、一応作成しておく)を作成する。
# code 29
enrich_1 <- original[, 1:59423] #
enrich_2 <- enrich_1 %>% rename("NAME" = "case")
enrich_3 <- enrich_2 %>% column_to_rownames("NAME")
enrich_3_t <- t(enrich_3) %>% data.frame()
enrich_3_t_1 <- enrich_3_t %>% rownames_to_column(var = "NAME") # this is for preparation of GCT file.
#
enrich_3_t_mat <-as.matrix(enrich_3_t) # this is for various enrichment analysis.
enrich_3_mat <- t(enrich_3) # this is for various enrichment analysis.
view(enrich_3_t_mat) # just checking the data.
view(enrich_3_mat) # just checking the data.
gc()
gc()準備が出来たので、cat()を使ってGCTファイル(要は上記の画像で示されるヤツ)を作成する。個人的に、cat()を使う方法が一番融通が菊のでは無いかと思う。必要な値としては、#1.2(これは固定値であり、変える必要はない)、遺伝子数(この解析では59422個)、症例数(この解析では10017例)、そして、各症例の遺伝子発現量のデータフレームenrich_3_t_1である。もちろん、ここではこれらの値をheader、number_gene、number_sampleといったオブジェクトに一旦入れている。そちらのほうが見やすいだろうから。それを順次、正規表現の改行”\n”をうまく使いながら、GCTファイルのフォーマット通りにcat()を使って入力しておく。cat()を使うときappend=TRUEにすることで、一つのファイルにどんどん追記していくことができる。逆にappend=FALSEになっていると、ファイルが上書きされてしまうのでGCTファイルが出来上がらない。ちなみに、append=TRUEにするとどんどん追記されていくので、GCTファイルを作成する前に、file.remove()を使って古いファイルを削除してから、改めて新しくファイルを作成したほうが混乱はない。諸々のヘッダーの情報をcat()で書き込んで行き、最後にwrite_tsv()で各症例における遺伝子発現量のデータフレームを書き込む。これでssGSEA2に使うGCTファイルの出来上がりである。
# code 30
header <- "#1.2"
number_gene <- nrow(enrich_3_t_1) # 59422 genes
number_sample <- ncol(enrich_3_t_1)-1 # 10017 cases
file.remove("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct")
cat(header, "\n", file = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct")
cat(number_gene, "\t", number_sample, "\n", file = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct", append = TRUE)
write_tsv(enrich_3_t_1, "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct",
col_names = TRUE,
append = TRUE)念のために出力したGCTファイルを読み込んで同じようなデータフレームが出来ているか確認しておく。間違っていたらssGSEA2のときにエラーが出るから、そのときに直せば良いのだが。
# code 31
check_ssGSEA2_gct <- read_tsv("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct", skip = 2)
症例毎にGSEAを行ってエンリッチメントスコアを計算する
いよいよGSEAに入る。この解析ではssGSEA2、escape、GVSA、fgseaを使用する。
ssGSEA2
これは、ここ(https://github.com/nicolerg/ssGSEA2)のパッケージを使用する。ソースコードを見る限りでは、ssGSEA2にはdoParallelが組み込まれており、par = 32と設定することで、32スレッドで計算出来るはずである。
ssGSEA2は計算にかなりの時間を要するので、どのくらい時間をかけたかを見るため、Sys.time()で開始時刻と終了時刻を記録しておき、最終的に費やした時間を計算するようにした。もう一つ、これも念のためではあるのが、何かしらの乱数を使っていると計算するごとに結果が微妙に違うということになるので、set.seed()でいつも同じシードを使うことにした。Broad instituteのGSEAではシードをいつも同じにしないと、計算が毎回微妙に違うはずである。
計算結果はresults_ssGSEA2_hallmark_NESオブジェクトとして保存する。全結果はカレントディレクトリ(もしくはホームディレクトリ)に保存されると思うので、必要ならばgetwd()で確認すると良い。
以下のコードは、GMTファイルとしてh.all.v2025.1.Hs.symbols.gmt(MsigDBのH: human hallmark gene sets。50個の代表的なヒトの分子パスウェイからなるGMTファイル。)を使用した場合のssGSEA2で、NES(Net Enrichment Score)を求めた場合のコードである。
# code 32
# registerDoParallel(cl = par, cores = 4)
start_results_ssGSEA2_hallmark_NES <- Sys.time()
set.seed(1)
results_ssGSEA2_hallmark_NES <- run_ssGSEA2(input.ds = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct",
output.prefix = "hallmark_NES_",
gene.set.databases = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/h.all.v2025.1.Hs.symbols.gmt",
sample.norm.type = "none",
weight = 0.75,
correl.type = "rank",
statistic = "area.under.RES",
output.score.type = "NES",
nperm = 1000,
min.overlap = 10,
extended.output = TRUE,
par = 32,
spare.cores = 0,
global.fdr = FALSE,
param.file=TRUE,
log.file='run.log_hallmark_NES')
end_results_ssGSEA2_hallmark_NES <- Sys.time()
gc()
gc()
# par = getOption("mc.cores",detectCores()) could be used.以下はssGSEA2によりES(Enrichmemt Score)を求めた場合である。
# code 33
start_results_ssGSEA2_hallmark_ES <- Sys.time()
set.seed(1)
results_ssGSEA2_hallmark_ES <- run_ssGSEA2(input.ds = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct",
output.prefix = "hallmark_ES_",
gene.set.databases = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/h.all.v2025.1.Hs.symbols.gmt",
sample.norm.type = "none",
weight = 0.75,
correl.type = "rank",
statistic = "area.under.RES",
output.score.type = "ES",
nperm = 1000,
min.overlap = 10,
extended.output = TRUE,
par = 32,
spare.cores = 0,
global.fdr = FALSE,
param.file=TRUE,
log.file='run.log_hallmark_ES')
end_results_ssGSEA2_hallmark_ES <- Sys.time()
gc()
gc()
# par = getOption("mc.cores",detectCores()) could be used.続いて、せっかく自分でHRRというGMTファイル(Homologous Recombination Repairの一つしかないけど…)も用意したので、それもやっておく。以下はNESを求めた場合。
# code 34
start_results_ssGSEA2_HRR_NES <- Sys.time()
set.seed(1)
results_ssGSEA2_HRR_NES <- run_ssGSEA2(input.ds = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct",
output.prefix = "HRR_NES_",
gene.set.databases = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/geneset_HRR.gmt",
sample.norm.type = "none",
weight = 0.75,
correl.type = "rank",
statistic = "area.under.RES",
output.score.type = "NES",
nperm = 1000,
min.overlap = 10,
extended.output = TRUE,
par = 32,
spare.cores = 0,
global.fdr = FALSE,
param.file=TRUE,
log.file='run.log_HRR_NES')
end_results_ssGSEA2_HRR_NES <- Sys.time()
gc()
gc()以下はGMTファイルとしてHRRを用いて、そのESを求めた場合。
# code 35
start_results_ssGSEA2_HRR_ES <- Sys.time()
set.seed(1)
results_ssGSEA2_HRR_ES <- run_ssGSEA2(input.ds = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/TCGA.gct",
output.prefix = "HRR_ES_",
gene.set.databases = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/geneset_HRR.gmt",
sample.norm.type = "none",
weight = 0.75,
correl.type = "rank",
statistic = "area.under.RES",
output.score.type = "ES",
nperm = 1000,
min.overlap = 10,
extended.output = TRUE,
par = 32,
spare.cores = 0,
global.fdr = FALSE,
param.file=TRUE,
log.file='run.log_HRR_ES')
end_results_ssGSEA2_HRR_ES <- Sys.time()
gc()
gc()escape
escapeは一般的にシングルセルRNAシークエンス(シングルセルトランスクリプトーム)のときに利用されるパッケージであるが、この症例数になってきたら、もはや同じような使い方が出来る(はずであるので、自分は平気で使っている。)。パッケージは個々にある(https://github.com/BorchLab/escape)。残念ながらescapeは並列化されていないようだが、そうでなくても十分に早い。ここからはGCTファイルではなく、通常のマトリックスenrich_3_t_matを使う。GMTファイルにもちょっと注意する必要がある。GMTファイルは各遺伝子セットを一つずつリストに入れなければならない。enrich_3_t_matはそもそもリストとして読み込まれているが、この時点ではHRRはリストではないので、上記で作成したgeneset_HRRをlist()に入れて、HRR_listを作成しておく。
# code 36
# escape, HRR
set.seed(1)
start_results_escape_HRR <- Sys.time()
HRR_list <- list(HRR = geneset_HRR)
# Version 2.0.0 <=
result_escape_HRR <- escape.matrix(
input.data = enrich_3_t_mat,
gene.sets = HRR_list,
method = "ssGSEA",
groups = 1000,
min.size = 10)
end_results_escape_HRR <- Sys.time()
gc()
gc()
# escape, hallmark
set.seed(1)
start_results_escape_hallmark <- Sys.time()
result_escape_hallmark <- escape.matrix(
input.data = enrich_3_t_mat,
gene.sets = hallmark_2025_1,
method = "ssGSEA",
groups = 1000,
min.size = 10)
end_results_escape_hallmark <- Sys.time()
gc()
gc()gsva
GSVAパッケージはここ(https://www.bioconductor.org/packages/release/bioc/html/GSVA.html)にある。これも特に並列化は組まれていないが、計算は十分に早い。escapeと同じように、遺伝子発現のマトリックスenrich_3_t_mat、GMTファイルのhallmark_2025_1とHRR_list(これはescapeのときにgeneset_HRRを使って改めて作成した)を使う。
# code 37
# gsva, HRR
set.seed(1)
start_results_GSVA_HRR <- Sys.time()
gsvaPar_HRR <- gsvaParam(enrich_3_t_mat, HRR_list)
result_gsva_HRR <- gsva(gsvaPar_HRR, verbose=TRUE)
end_results_GSVA_HRR <- Sys.time()
gc()
gc()
# geva, hallmark
set.seed(1)
start_results_GSVA_hallmark <- Sys.time()
gsvaPar_hallmark <- gsvaParam(enrich_3_t_mat, hallmark_2025_1)
result_gsva_hallmark <- gsva(gsvaPar_hallmark, verbose=TRUE)
end_results_GSVA_hallmark <- Sys.time()
gc()
gc()fgsea
これもシングルセルRNAシークエンス(シングルセルトランスクリプトーム)でよく使用されるパッケージで、ここ(https://bioconductor.org/packages/release/bioc/html/fgsea.html)にある。これはescapeやgsvaのように全症例を一気に、マトリックスとして計算してくれないのでちょっと面倒くさい。その一方で、他の解析やコードにうまく組くことが出来るという柔軟性も持っているような気がする。ただし、そもそも大きいデータセットを計算するので、システムの性能が追いつけば、の話ではあるが。この問題については後ほど少し述べる。
fgseaは、症例毎に結果のオブジェクトが出てきてしまう。今回の解析では症例数が10017例、遺伝子セットがh.all.v2025.1.Hs.symbols.gmtを用いる場合は50個あるので、下手すると大量のオブジェクトが出力されてしまう。そんなことになったら面倒くさいのは確実なので、出力はリストに入れることにする。
以下は遺伝子セットとしてHRRを使った場合である。fgsea()の結果を事前に準備したresult_fgesa_HRRリストにいれて、そこから欲しいデータを各データフレーム(result_fgsea_ES_HRR、result_fgsea_NES_HRR、result_fgsea_padj_HRR)に入れていく。これらは、fsgea()の前に空のデータフレームを用意しておくと、エラーがでない。最終的に出来上がった各データフレームには、rawnames()とcolnames()で列名と行名を付けておくほうが、後々混乱せずに済む。
そして、以下は時間をかけてfor()で各症例のfgsea()を計算する場合である。後ほどforeach()を使って高速化した方法を記すが、残念ながら、このfor()を使った方法がクラッシュしない一番安全な正攻法である。
# code 38
# prepare geneset for fgsea, HRR, single thread
geneset_fgsea_HRR <- list()
geneset_fgsea_HRR[[1]] <- geneset_HRR
names(geneset_fgsea_HRR) <- "HRR"
# fgsea, HRR
result_fgesa_HRR <- list()
result_fgsea_ES_HRR <- data.frame()
result_fgsea_NES_HRR <- data.frame()
result_fgsea_padj_HRR <- data.frame()
start_results_fgsea_HRR <- Sys.time()
for(i in 1:ncol(enrich_3_t_mat)) {
result_fgesa_HRR[[i]] <- fgsea(pathways = geneset_fgsea_HRR,
stats = enrich_3_t_mat[,i],
minSize = 10,
maxSize = 500)
result_fgsea_ES_HRR[1:length(geneset_fgsea_HRR),i] <- result_fgesa_HRR[[i]]$ES
result_fgsea_NES_HRR[1:length(geneset_fgsea_HRR),i] <- result_fgesa_HRR[[i]]$ES
result_fgsea_padj_HRR[1:length(geneset_fgsea_HRR),i] <- result_fgesa_HRR[[i]]$padj}
end_results_fgsea_HRR <- Sys.time()
# Enrichment Score; ES, HRR
colnames(result_fgsea_ES_HRR) <- colnames(enrich_3_t_mat)
rownames(result_fgsea_ES_HRR) <- names(geneset_fgsea_HRR)
# Normalized Enrichment Score; NES, HRR
colnames(result_fgsea_NES_HRR) <- colnames(enrich_3_t_mat)
rownames(result_fgsea_NES_HRR) <- names(geneset_fgsea_HRR)
# FDR adjusted p-value; padj, HRR
colnames(result_fgsea_padj_HRR) <- colnames(enrich_3_t_mat)
rownames(result_fgsea_padj_HRR) <- names(geneset_fgsea_HRR)
gc()
gc()以下が、for()をforeach()に変えたコードである。遺伝子セットが1つだけなので、余裕で計算できる。
# code 39
# # fgsea using foreach, HRR, multi treads
# # prepare geneset for fgsea, msigdb,
# geneset_fgsea <- fgsea::gmtPathways("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/msigdb_v2025.1.Hs_files_to_download_locally/msigdb_v2025.1.Hs_GMTs/msigdb.v2025.1.Hs.symbols.gmt")
# prepare geneset for fgsea, HRR
geneset_fgsea_HRR <- list()
geneset_fgsea_HRR[[1]] <- geneset_HRR
names(geneset_fgsea_HRR) <- "HRR"
# register threads
n_workers <- max(1, detectCores(logical = FALSE) - 1)
cl <- makeCluster(n_workers, type = "PSOCK")
registerDoParallel(cl)
# # prevent thread oversubscription for each thread
# clusterEvalQ(cl, {
# Sys.setenv(OMP_NUM_THREADS="1",
# MKL_NUM_THREADS="1",
# OPENBLAS_NUM_THREADS="1",
# RCPP_PARALLEL_NUM_THREADS="1")})
# # prevent thread oversubscription for each thread
# # this can also complete calculation.
# # fgsea was able to be run by means of adding environmental variables(su; cd /etc; gedit environment) on the /etc/environment file.
clusterEvalQ(cl, {
Sys.setenv(OMP_NUM_THREADS="1",
OMP_THREAD_LIMIT="1",
OMP_MAX_ACTIVE_LEVELS="1",
OMP_DYNAMIC=FALSE,
MKL_NUM_THREADS="1",
MKL_DYNAMIC=FALSE,
OPENBLAS_NUM_THREADS="1",
GOTO_NUM_THREADS="1" ,
RCPP_PARALLEL_NUM_THREADS="1")})
start_results_fgsea_foreach_HRR <- Sys.time()
set.seed(1)
result_fgesa_foreach_HRR <- list()
result_fgesa_foreach_HRR <- foreach(i = seq_len(ncol(enrich_3_t_mat)), .packages = "fgsea", .inorder = TRUE) %dopar% {
fgsea(pathways = geneset_fgsea_HRR,
stats = enrich_3_t_mat[, i],
minSize = 10,
maxSize = 500)}
stopCluster(cl)
foreach::registerDoSEQ()
end_results_fgsea_foreach_HRR <- Sys.time()
# extruct results of fgsea, foreach, HRR
result_fgesa_foreach_ES_HRR <- data.frame()
result_fgesa_foreach_NES_HRR <- data.frame()
result_fgesa_foreach_padj_HRR <- data.frame()
for(i in 1:length(result_fgesa_foreach_HRR)){
result_fgesa_foreach_ES_HRR[1:length(geneset_fgsea_HRR),i] <- data.frame(result_fgesa_foreach_HRR[[i]]$ES)
result_fgesa_foreach_NES_HRR[1:length(geneset_fgsea_HRR),i] <- data.frame(result_fgesa_foreach_HRR[[i]]$NES)
result_fgesa_foreach_padj_HRR[1:length(geneset_fgsea_HRR),i] <- data.frame(result_fgesa_foreach_HRR[[i]]$padj)
}
rownames(result_fgesa_foreach_ES_HRR) <- "HRR"
colnames(result_fgesa_foreach_ES_HRR) <- colnames(enrich_3_t_mat)
rownames(result_fgesa_foreach_NES_HRR) <- "HRR"
colnames(result_fgesa_foreach_NES_HRR) <- colnames(enrich_3_t_mat)
rownames(result_fgesa_foreach_padj_HRR) <- "HRR"
colnames(result_fgesa_foreach_padj_HRR) <- colnames(enrich_3_t_mat)そして、以下が50個の遺伝子セットを使ってforeach()を使ってfgsea()を計算する方法である。ここで問題が起こった。別にポストしたのだが、どうやらSMT(Simoltanous Multi Threading)が上手く動いていないことが判明した。自分は今Ryzen 9950Xを使っているが、intel core i9とか使っているときはこんなことなかったように思うのだが….ということで、まずは並列化のオーバーレジストレーションをさせないために、Sys.setenv()で割り当てていくスレッドを1つずつに限定する。これで、この計算は上手くいった。後は、上記と同じように出てきた結果のうち、欲しい値(NES、ES、padj)をデータフレームに入れておく。
# code 40
# fgsea using foreach, hallmark, multi threads
# prepare geneset for fgsea, hallmark
geneset_fgsea_hallmark <- fgsea::gmtPathways("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/h.all.v2025.1.Hs.symbols.gmt")
# # prepare geneset for fgsea, msigdb
# geneset_fgsea <- fgsea::gmtPathways("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/msigdb_v2025.1.Hs_files_to_download_locally/msigdb_v2025.1.Hs_GMTs/msigdb.v2025.1.Hs.symbols.gmt")
# # prepare geneset for fgsea, HRR
# geneset_fgsea_HRR <- list()
# geneset_fgsea_HRR[[1]] <- geneset_HRR
# names(geneset_fgsea_HRR) <- "HRR"
# From chatGPT
# choose workers (physical cores minus one is a good start)
n_workers <- max(1, detectCores(logical = FALSE) - 1)
cl <- makeCluster(n_workers, type = "FORK") #FORK #PSOCK
registerDoParallel(cl)
# # prevent thread oversubscription inside workers (optional but recommended)
# clusterEvalQ(cl, {
# Sys.setenv(OMP_NUM_THREADS="1",
# MKL_NUM_THREADS="1",
# OPENBLAS_NUM_THREADS="1",
# RCPP_PARALLEL_NUM_THREADS="1")})
# # prevent thread oversubscription inside workers (optional but recommended)
# # this can also complete calculation.
# # fgsea was able to be run by means of adding environmental variables(su; cd /etc; gedit environment) on the /etc/environment file.
clusterEvalQ(cl, {
Sys.setenv(OMP_NUM_THREADS="1",
OMP_THREAD_LIMIT="1",
OMP_MAX_ACTIVE_LEVELS="1",
OMP_DYNAMIC=FALSE,
MKL_NUM_THREADS="1",
MKL_DYNAMIC=FALSE,
OPENBLAS_NUM_THREADS="1",
GOTO_NUM_THREADS="1" ,
RCPP_PARALLEL_NUM_THREADS="1")})
start_results_fgsea_foreach_hallmark <- Sys.time()
set.seed(1)
result_fgesa_foreach_hallmark <- list()
result_fgesa_foreach_hallmark <- foreach(i = seq_len(ncol(enrich_3_t_mat)), .packages = "fgsea", .inorder = TRUE) %dopar% {
fgsea(pathways = geneset_fgsea_hallmark,
stats = enrich_3_t_mat[, i],
minSize = 10,
maxSize = 500)}
stopCluster(cl)
foreach::registerDoSEQ()
# Extract results of fgsea, foreach, hallmark
result_fgesa_foreach_ES_hallmark <- data.frame()
result_fgesa_foreach_NES_hallmark <- data.frame()
result_fgesa_foreach_padj_hallmark <- data.frame()
for(i in 1:length(result_fgesa_foreach_hallmark)){
result_fgesa_foreach_ES_hallmark[1:length(geneset_fgsea_hallmark),i] <- data.frame(result_fgesa_foreach_hallmark[[i]]$ES)
result_fgesa_foreach_NES_hallmark[1:length(geneset_fgsea_hallmark),i] <- data.frame(result_fgesa_foreach_hallmark[[i]]$NES)
result_fgesa_foreach_padj_hallmark[1:length(geneset_fgsea_hallmark),i] <- data.frame(result_fgesa_foreach_hallmark[[i]]$padj)
}
colnames(result_fgesa_foreach_ES_hallmark) <- colnames(enrich_3_t_mat)
rownames(result_fgesa_foreach_ES_hallmark) <- names(geneset_fgsea_hallmark)
colnames(result_fgesa_foreach_NES_hallmark) <- colnames(enrich_3_t_mat)
rownames(result_fgesa_foreach_NES_hallmark) <- names(geneset_fgsea_hallmark)
colnames(result_fgesa_foreach_padj_hallmark) <- colnames(enrich_3_t_mat)
rownames(result_fgesa_foreach_padj_hallmark) <- names(geneset_fgsea_hallmark)
end_results_fgsea_foreach_hallmark <- Sys.time()fgsea使用時の注意点
ちょっとここで、重要なことがある。fgsea()はBiocParalelを使った並列化が組み込まれている。こういう場合にさらにdoParallelを重ねたりすると、今度はこのようにSMTは上手くいかないし、割り当てるデータが大きくなりすぎてどうやってもメモリリークみたいなことになってしまう。今回は遺伝子セットが50個だけなのでメモリリーク的なことで落ちることはなかったが、これがmsigdb.v2025.1.Hs.symbols.gmtのように35000個もあるような場合は、家庭用のコンピューターでは無理な気がする。第一、MsigDBに「遺伝子セット全部使ったエンリッチメント解析なんて、やるなよ!!」と以下の画像の通り書いてある。その通りである。正直、出来るような気はするのだが、計算出来たところで、それが正しく計算出来ているか検証する必要もありそうである。時間が十分ある場合はやっても良さそうだが、別の方法を探ったほうが良いかもしれない。
ssGSEA2・escape・GSVA・fgseaの計算時間のまとめ
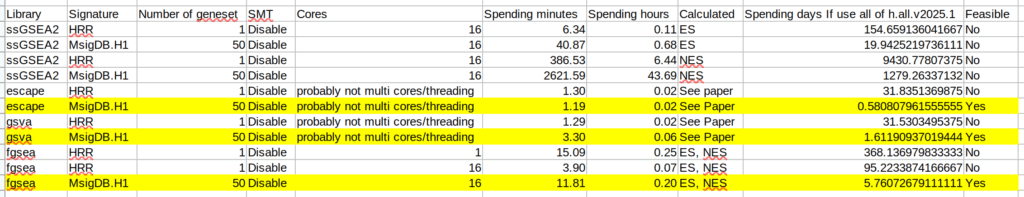
以下の表が、各パッケージを用いた計算に要した時間である。一番右の列にあるのが、もしMsigDBに登録のあるヒトの遺伝子セット全部を一気にエンリッチメント解析したときの各パッケージの推定計算時間である。ssGSEA2なんて、1200日以上必要らしい。全く現実的ではない。fgseaも、foreachで上手く並列処理できれば、1週間くらいで終わるが、そのためは計算方法を工夫したり、コンピューターをうまいこと調整(?良く知らんが)しなければならないようだ。そこまでしてfgsea使うかって感じではある。これが、遺伝子セットは選んで計算しろ、ということだろう。現実的なのはescape、gsvaである。
各種GSEAの結果をまとめる
計算結果は一つのオブジェクトにまとまっている方が、後の解析でも便利だと思う。ということで、各GSEAの結果を一つのオブジェクトにまとめることにする。解析の順番はssGSEA2が最初だったが、このssGSEA2の結果だけ他と違うように保存されているため、まずはescap、GSVA、fgseaをまとめて、最後にssGSEA2をまとめる。なんか長々と書いてあるけど、やっていること自体は何も難しくない。エクセルでGUI(Graphical User Interface)でまとめるのと変わらない。それがCUI(Console User Interface)になっただけである。
escape
escapeの結果は、行が症例(case1 からcase10017)、列がescapeのエンリッチメントスコアとして出力される。マトリックスresult_escape_hallmarkがそれである。
ここで、各GSEAに使用したGMTファイルは同じものであり、結果の列名もしくは行名が各GSEAの結果で同じになっているはずである。これでは最後に結合したときにどの解析で得られたエンリッチメントスコアなのかわからないので、各遺伝子セットの末尾に使用したパッケージの名前(escape、gsva、fgsea、ssGSEA2のどれか)を付けることにする。
そのためには、まずは列名(各遺伝子セット名が付いている)をcolnames()で取り出してきて、それを1列だけのデータフレームcolumn_escape_hallmarkとして保存し、その列名にgenesetという名前を付ける。そして、そのデータフレームcolumn_escape_hallmarkにmutate()で列libraryを追加し、それにescapeという値を入れ、列genesetと列libraryをunite()で結合して、新しくgenesetという列にする。それをcolnames()を使って改めてマトリックスresult_escape_hallmarkの列名にする。同様にマトリックスresult_escape_HRRにもこの処理をする。
次にマトリックスresult_escape_hallmarkとマトリックスresult_escape_HRRをas.data.frame()でデータフレームに直し、rownames_to_column()で行名である各症例の通し番号を列caseにする。そしてその両者のデータフレームを列名caseをメインキー(軸)にしてinner_joinする。
# code 41
# escape
## prepare column name, hallmark
column_escape_hallmark <- colnames(result_escape_hallmark) %>% data.frame()
colnames(column_escape_hallmark) = "geneset"
column_escape_hallmark <- column_escape_hallmark %>% mutate(library = "escape")
column_escape_hallmark <- column_escape_hallmark %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_escape_hallmark) <- column_escape_hallmark$geneset
## prepare column name, HRR
column_escape_HRR <- colnames(result_escape_HRR) %>% data.frame()
colnames(column_escape_HRR) = "geneset"
column_escape_HRR <- column_escape_HRR %>% mutate(library = "escape")
column_escape_HRR <- column_escape_HRR %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_escape_HRR) <- column_escape_HRR$geneset
## hallmark
result_escape_hallmark_1 <- as.data.frame(result_escape_hallmark) %>% rownames_to_column(var = "case")
## HRR
result_escape_HRR_1 <- as.data.frame(result_escape_HRR) %>% rownames_to_column(var = "case")
## merge hallmark and HRR
result_escape <- inner_join(result_escape_hallmark_1, result_escape_HRR_1, by = "case")GSVA
GSVAの出力結果はescapeとは違い、行がGSVAによるエンリッチメントスコア、列が症例(case1 からcase10017)になる。なので、最初にt()で行列を入れ替えて、そこからescapeと同じような手順で処理をしていく。
# code 42
# gsva
## prepare dataset
result_gsva_hallmark_t <- result_gsva_hallmark %>% t() %>% data.frame()
result_gsva_HRR_t <- result_gsva_HRR %>% t() %>% data.frame()
## prepare column name, hallmark
column_gsva_hallmark <- colnames(result_gsva_hallmark_t) %>% data.frame()
colnames(column_gsva_hallmark) = "geneset"
column_gsva_hallmark <- column_gsva_hallmark %>% mutate(library = "gsva")
column_gsva_hallmark <- column_gsva_hallmark %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_gsva_hallmark_t) <- column_gsva_hallmark$geneset
## prepare column name, HRR
column_gvsa_HRR <- colnames(result_gsva_HRR_t) %>% data.frame()
colnames(column_gvsa_HRR) = "geneset"
column_gvsa_HRR <- column_gvsa_HRR %>% mutate(library = "gsva")
column_gvsa_HRR <- column_gvsa_HRR %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_gsva_HRR_t) <- column_gvsa_HRR$geneset
## inner_join by case
result_gsva_hallmark_1 <- result_gsva_hallmark_t %>% rownames_to_column(var = "case")
result_gsva_HRR_1<- result_gsva_HRR_t %>% rownames_to_column(var = "case")
result_gsva <- inner_join(result_gsva_hallmark_1, result_gsva_HRR_1, by = "case")
fgsea
fgseaの出力は各遺伝子セット毎に結果が出てきてしまい、非常に面倒なので、既に上記で行がfgseaによるエンリッチメントスコア、列が症例(case1 からcase10017)のデータフレームに直している。そのとき、NES(Normalized Enrichment Score)、ES(Enrichment Score)、padj(FDR補正後のp値;FDR adjusted p value)をそれぞれ作成しているが、ここではNESを使う。これもGSVAと同じように処理すれば良い。
# code 43
# fgsea
## prepare dataset
result_fgesa_foreach_NES_hallmark_t <- result_fgesa_foreach_NES_hallmark %>% t() %>% data.frame()
result_fgesa_foreach_NES_HRR_t <- result_fgesa_foreach_NES_HRR %>% t() %>% data.frame()
## prepare column name, hallmark
column_fgsea_hallmark <- colnames(result_fgesa_foreach_NES_hallmark_t) %>% data.frame()
colnames(column_fgsea_hallmark) = "geneset"
column_fgsea_hallmark <- column_fgsea_hallmark %>% mutate(library = "fgsea")
column_fgsea_hallmark <- column_fgsea_hallmark %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_fgesa_foreach_NES_hallmark_t) <- column_fgsea_hallmark$geneset
## prepare column name, HRR
column_fgsea_HRR <- colnames(result_fgesa_foreach_NES_HRR_t) %>% data.frame()
colnames(column_fgsea_HRR) = "geneset"
column_fgsea_HRR <- column_fgsea_HRR %>% mutate(library = "fgsea")
column_fgsea_HRR <- column_fgsea_HRR %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_fgesa_foreach_NES_HRR_t) <- column_fgsea_HRR$geneset
## inner_join by case
result_fgesa_foreach_NES_hallmark_1 <- result_fgesa_foreach_NES_hallmark_t %>% rownames_to_column(var = "case")
result_fgesa_foreach_NES_HRR_1 <- result_fgesa_foreach_NES_HRR_t %>% rownames_to_column(var = "case")
result_fgsea <- inner_join(result_fgesa_foreach_NES_hallmark_1, result_fgesa_foreach_NES_HRR_1, by = "case")
ssGSEA2
初見殺しなのはssGSEA2の結果である。これはもちろんオブジェクトでも保存するのだが、なんと最終的にはGCTファイルが作業ディレクトリに出力される。非常にやりにくい。末尾が”-score.gct”のファイルが必要なファイルである。しかも、プロテオミクスにも対応しているようなフォーマットのGCTファイルらしく、頭の行が#1.2ではなく、#1.3となっている。さらに、これにはエンリッチメント比(Overrepresentation analysisのそれ)や、エンリッチされてきた遺伝子が書かれた列も入っている。これはこれで有用なのだが、ここでは着目しない。
GCTファイルを読み込んだら、上記の不要な列を削除し、fgseaやGSVAの出力結果と同じようなフォーマットのデータフレームにする。それ以降はfgseaやGSVAと同様に処理する。
# code 44
# ssGSEA2
## prepare dataset
result_ssGSEA2_hallmark_gct_NES <- read_tsv("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/hallmark_NES_-scores.gct", skip = 2)
result_ssGSEA2_HRR_gct_NES <- read_tsv("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/HRR_NES_-scores.gct", skip = 2)
result_ssGSEA2_hallmark_gct_NES <- result_ssGSEA2_hallmark_gct_NES[,-(2:20038)]
result_ssGSEA2_HRR_gct_NES <- result_ssGSEA2_HRR_gct_NES[,-(2:20038)]
result_ssGSEA2_hallmark_gct_NES <- result_ssGSEA2_hallmark_gct_NES %>% column_to_rownames(var = "id")
result_ssGSEA2_HRR_gct_NES <- result_ssGSEA2_HRR_gct_NES %>% column_to_rownames(var = "id")
colnames(result_ssGSEA2_hallmark_gct_NES) <- original$case
colnames(result_ssGSEA2_HRR_gct_NES) <- original$case
result_ssGSEA2_hallmark_gct_NES_t <- result_ssGSEA2_hallmark_gct_NES %>% t() %>% data.frame()
result_ssGSEA2_HRR_gct_NES_t <- result_ssGSEA2_HRR_gct_NES %>% t() %>% data.frame()
## prepare column name, hallmark
column_ssGSEA2_hallmark <- colnames(result_ssGSEA2_hallmark_gct_NES_t) %>% data.frame()
colnames(column_ssGSEA2_hallmark) = "geneset"
column_ssGSEA2_hallmark <- column_ssGSEA2_hallmark %>% mutate(library = "ssGSEA2")
column_ssGSEA2_hallmark <- column_ssGSEA2_hallmark %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_ssGSEA2_hallmark_gct_NES_t) <- column_ssGSEA2_hallmark$geneset
## prepare column name, HRR
column_ssGSEA2_HRR <- colnames(result_ssGSEA2_HRR_gct_NES_t) %>% data.frame()
colnames(column_ssGSEA2_HRR) = "geneset"
column_ssGSEA2_HRR <- column_ssGSEA2_HRR %>% mutate(library = "ssGESA2")
column_ssGSEA2_HRR <- column_ssGSEA2_HRR %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_ssGSEA2_HRR_gct_NES_t) <- column_ssGSEA2_HRR$geneset
## inner_join by case
result_ssGSEA2_hallmark_gct_NES_t_1 <- result_ssGSEA2_hallmark_gct_NES_t %>% rownames_to_column(var = "case")
result_ssGSEA2_HRR_gct_NES_t_1 <- result_ssGSEA2_HRR_gct_NES_t %>% rownames_to_column(var = "case")
result_ssGSEA2 <- inner_join(result_ssGSEA2_hallmark_gct_NES_t_1, result_ssGSEA2_HRR_gct_NES_t_1, by = "case")最後に結合したいデータフレームを一つずつリストに入れて、それをreduce()で一気にinner_join()する。最終的に出来上がるデータフレームanalysisには、遺伝子発現量と各GSEAのパッケージにより求めたエンリッチメントスコアの値が入っている。これを使って症例を階層化し、どのような遺伝子もしくは遺伝子セットが患者の生存率に統計的有意な影響を及ぼすかを解析する。
このようなサイズの大きなデータフレームやマトリックスの処理は、メモリをかなり消費する可能性があるので、コンピューターがクラッシュしないように様子をみてやったほうが良いと思う。対策としては、少なくともSWAPを十分に確保しておく必要がある。また、SWAPを活用して解析している場合は、途中からなんだかinner_join()が遅いということがあると思う。おそらくそれはスワップスラッシングというヤツだと思う。気長に待つか、それが出来ない場合は何らかの対策が必要である。すでに必要なデータをH5DFで保存しているわけで、この解析をやっているとき何度Pythonに逃げようとしたことか…そして、「いや〜今からPythonってなぁ….ここまでやってしまったからRのでやるか…」という感じである。
本当に、このメモリ不足はRの敵である。 メモリがいくらあっても足りない。これって、大きいサーバーとかならこういったメモリ不足とかどうしてるんだろうか。勝手にこういった解析を流したら、管理者に怒られるのだろうか。
# code 45
# merge all dataset
temp <- list()
temp[[1]] <- original
temp[[2]] <- result_escape
temp[[3]] <- result_fgsea
temp[[4]] <- result_gsva
temp[[5]] <- result_ssGSEA2
analysis <- reduce(temp, inner_join) %>% data.frame()
view(analysis[,59494:59697])
疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する
ようやっと解析出来る….ここまでは本当にストレスだった。解析なんかなにもしていない。データフレームをクリーニングしたり結合したりフィルターかけたり、そればっかり。
早速、症例毎の遺伝子発現量や遺伝子セットのエンリッチメントスコアをまとめたデータフレームanalysisを使って、解析を行う。
まずは、適当にヒストグラムを表示してみる。ここではGSVAによって求めた遺伝子セットHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaを見てみることにする。ついでに、median()、range()、quantile()で、どんな感じの分布(というかどんな範囲の値)なのかを確認してみる。出来たグラフはggsave()で保存する。ggsave()は直前にggplot2を使って描かれた図を保存出来る。
# code 46
# histogram
# gvsa
ggplot(analysis, aes(x = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
geom_histogram(binwidth = 10*(1/nrow(analysis))) +
xlim(range(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva))
# check the range of values
median(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)
range(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)
quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75))
# output
ggsave("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/histogram_HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva.png")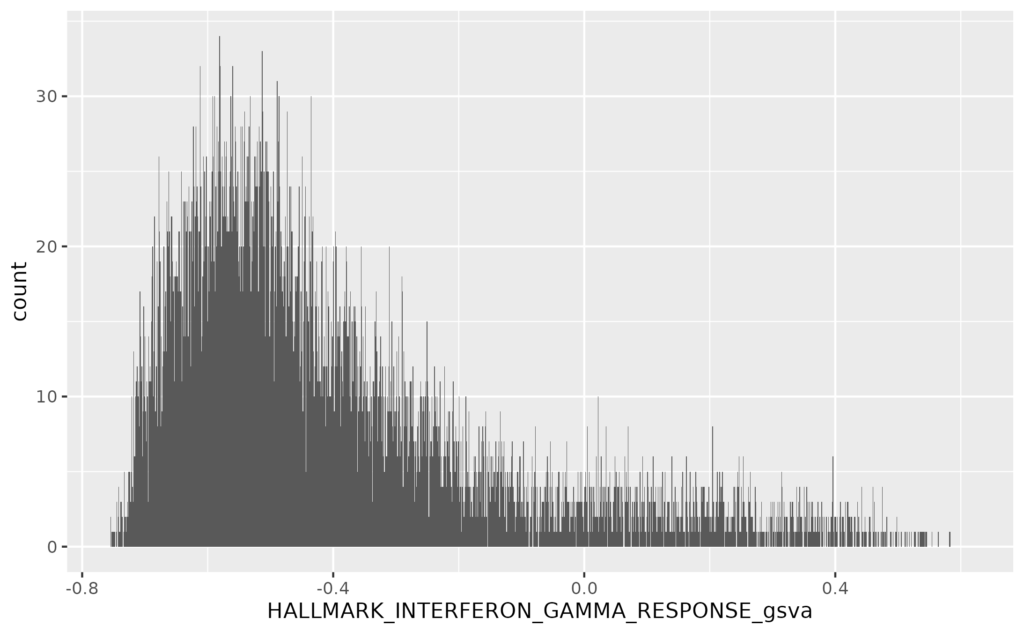
せっかくなので、fgseaによって求めた遺伝子セットHALLMARK_INTERFERON_GAMMA_RESPONSE_fgseaも同様に見てみる。
# code 47
# histogram
# fgsea
ggplot(analysis, aes(x = HALLMARK_INTERFERON_GAMMA_RESPONSE_fgsea)) +
geom_histogram(binwidth = 10*(1/nrow(analysis))) +
xlim(range(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_fgsea))
# check the range of values
median(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_fgsea)
range(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_fgsea)
quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_fgsea, probs = c(0.25, 0.5, 0.75))
# output
ggsave("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/histogram_HALLMARK_INTERFERON_GAMMA_RESPONSE_fgsea.png")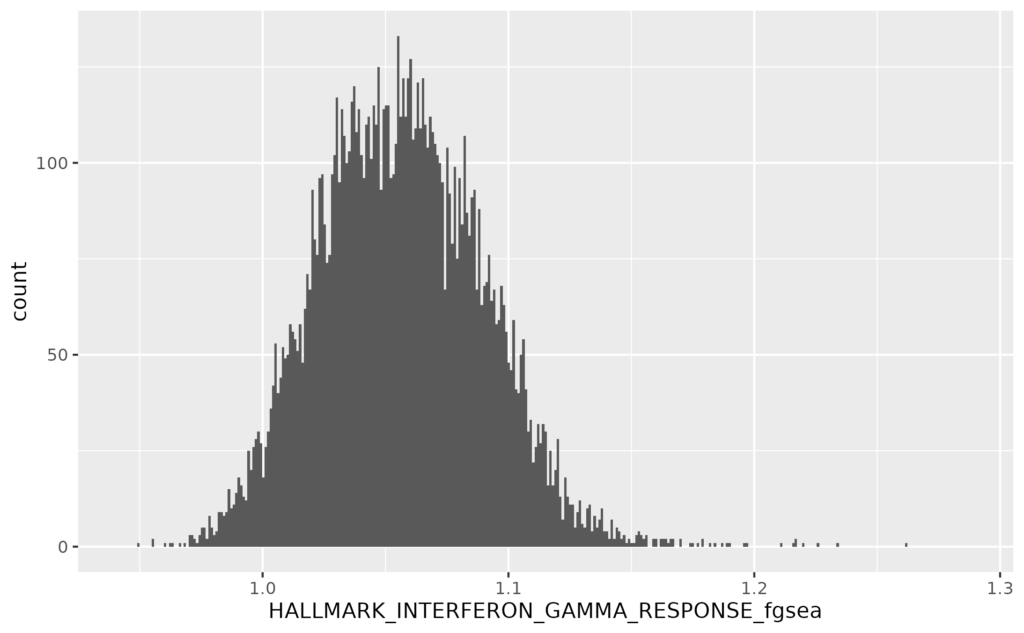
なんか、GSVAの結果のほうが階層に別れてそうで解析しやすいかも、ということがわかる。違いを示すのは大事である。そういうことなので、以降はGSVAの結果を階層化に使用することにする。
疾患毎の遺伝子発現量やエンリッチメントスコアをバイオリンプロットで比較する
ヒストグラムでは、明確なピークが複数出現する、みたいなことは無いだろうと思う。それに、上記のヒストグラムでは何かと何かを比較していない。ある遺伝子もしくは遺伝子セットが、どのような範囲で発現しているかくらいしかわからない。何かを見つけようとする場合、何かを基準値と比較する必要がある。そしてこれはヒトのデータであり、各値がどんな分布に従うのかは未知である。
こういったデータの比較には、バイオリンプロットがやはり良いだろう。ということで、バイオリンプロットにより疾患毎にある特定の遺伝子もしくは遺伝子セットを比較する。
バージョン1;とりあえずバイオリンプロットを表示する
言うて、関心のある遺伝子もしくは遺伝子セットがどんな感じにバイオリンプロットで表示されるのかは、今の所よくわからない。なので、とりあえずバイオリンプロットを出力してみようと思う。ここではGSVAによるエンリッチメントスコアHALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION_gsvaに着目する。そして、横軸に列diseaseを、縦軸に列HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION_gsvaを取る。列diseaseにはBRCAとかLUADとかの疾患名が保持されている。したがって、このバイオリンプロットでは、疾患毎のHALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITIONのエンリッチメントスコアを表示する。バイオリンプロットでも生存率でも、基本的にはggplot2を利用する。これは基本的に比較的キレイな図がかけるためである。キレイな図がかけるパッケージとしては、ggplot2の他にはplotlyなんかもある。
図の保存は、上記のヒストグラムのようにggsave()を使うのももちろん良いが、自分はより細かく設定できるRの基本的なパッケージに入っているtiff()を使うのが好きである。
# code 48
# violin plot, version 1
## gvsa
### If log10 scale is better for y-axis, use "scale_y_log10() +" before "theme()".
ggplot(analysis, aes(x = disease, y = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
geom_violin(width = 1.2, trim = FALSE, fill = "lightblue") +
ylim(range(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
theme(axis.text.x = element_text(angle = 90, hjust = 1))
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/","HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva","_v1", ".tiff", sep = "")
tiff(filename = path,
width = 1200, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
ggplot(analysis, aes(x = disease, y = HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION_gsva)) +
geom_violin(width = 1.2, trim = FALSE, fill = "lightblue") +
ylim(range(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
theme(axis.text.x = element_text(angle = 90, hjust = 1))
dev.off()以下のようなバイオリンプロットが表示された。どの疾患でエンリッチメントスコアHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaが高いとか低いとか、なんとなくわかる、気がする。
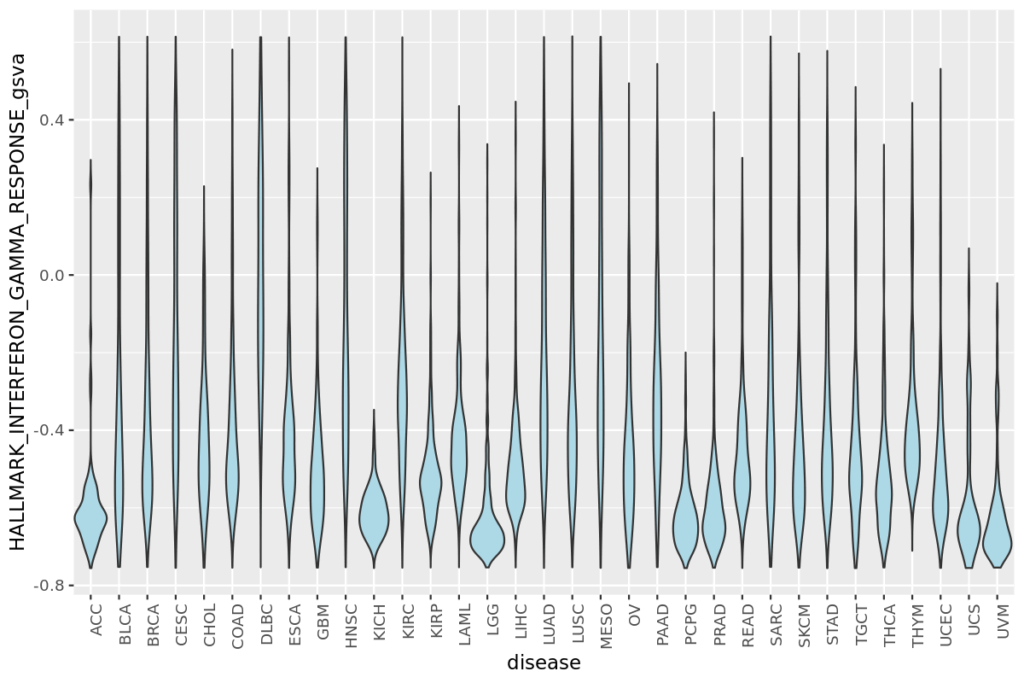
バージョン2;疾患全体の中央値(50パーセンタイル)、25パーセンタイル、75パーセンタイルを黒、青、赤の水平線で表示する
少なくとも中央値、75パーセンタイル、25パーセンタイルくらいは表示したら、どの疾患でより高いか、低いかが見やすくなるような気がする。なので、それらを水平線で入れることにする。
ggplot2の特徴は、こうやって後から付け足すことが出来る点である。レイヤーを追加するという言い方をするらしい。なんかこれってggplot2の特徴とか言われるけど、R の基本的な描画パッケージも、append = TRUEとかで追加していけるような気がするが…まあいい。
そのためにはgeom_hline()を使う。その中で、quantile()を使って全体のHALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION_gsvaの中央値、25パーセンタイル、75パーセンタイルを計算させ、それをそれぞれ黒、青、赤の水平線で入れる。
# code 49
# violong plot, version 2
## gvsa
### If log10 scale is better for y-axis, use "scale_y_log10() +" before "theme()".
ggplot(analysis, aes(x = disease, y = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
geom_violin(width = 1.2, trim = FALSE, fill = "lightblue") +
geom_hline(yintercept = quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
theme(axis.text.x = element_text(angle = 90, hjust = 1))
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/","HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva","_v2", ".tiff", sep = "")
tiff(filename = path,
width = 1200, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
ggplot(analysis, aes(x = disease, y = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
geom_violin(width = 1.2, trim = FALSE, fill = "lightblue") +
geom_hline(yintercept = quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
theme(axis.text.x = element_text(angle = 90, hjust = 1))
dev.off()以下が出力である。なんとなく疾患毎の差が見やすくなった、気がする。いや、ちがう。一目瞭然のヤツが欲しい。
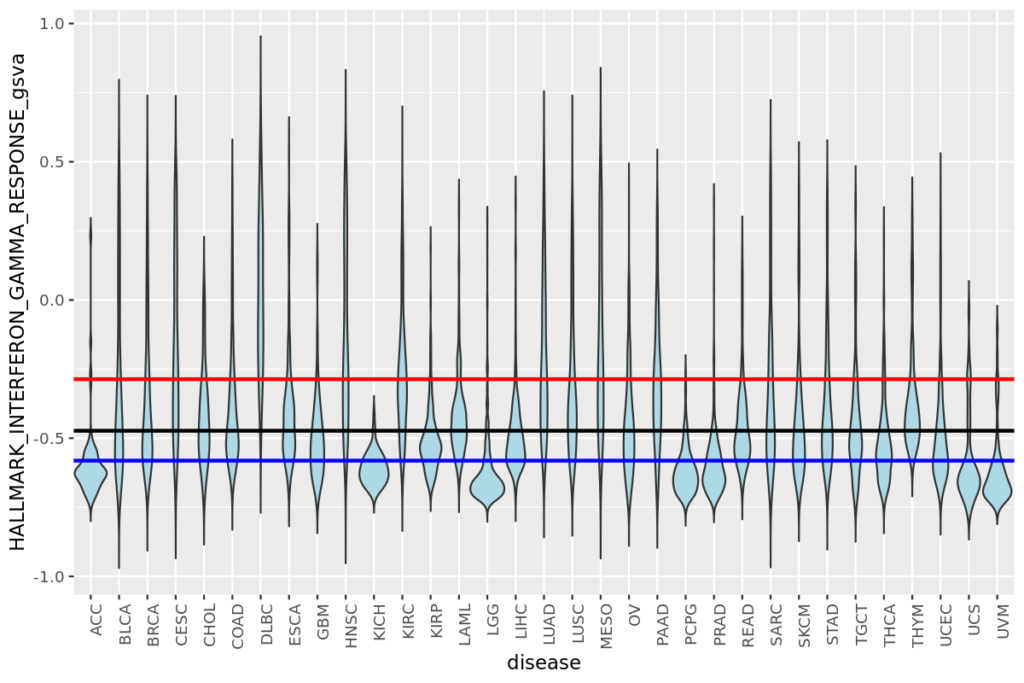
バージョン3;疾患毎の中央値が、全体の中央値の1.5倍以上、0.75以下、それ以外の疾患を、それぞれピンク、水色、グレーで表示する
一目瞭然というのは、サイエンスでも重要である。一目瞭然でない場合は、変なヤツがグダグダ言ってるだけにしか見えない。一目瞭然でないということは、極論を言えば、わからない聴衆が悪い、ということを聴衆に強いていると見なすことも可能であり、そんなプレゼンテーションは成功であるはずがない。それに、このような邪魔くさいデータではなおさら見やすくする努力は必要だろうと思う。
見やすくするためには、見てほしいものを目立たせる必要がある。ここでは、他に比べてHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaの値が高い疾患をピンクで、低い疾患を青で、全体の中央値と変わらないものはグレーで表示することにする。
では、高いとか低いとかをどうやって決めるか。ここでは、各疾患の中央値が、全体の75パーセンタイル以上の場合はピンク、全体の25パーセンタイル以下ならば青、25パーセンタイルより高く75パーセンタイルより低いならばグレーにした。統計については色々やったけど、例えば、対象とする疾患以外の全疾患との有意差をwilcox.test()したりとかやったけど、この処理は互いに独立なのか、比較する上での症例数がマッチしているのかなど、いまいち正しい確証がなかったので、こういったシンプルな方法に落ち着けた。
# code 50
# violin plot version 3
## gvsa
Q50_all_v3 <- median(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva) # Q50_all_EMT = -0.4754158
Q25_all_v3 <- quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = 0.25) # Q25_all_EMT = -0.5745684
Q75_all_v3 <- quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = 0.75) # Q75_all_EMT = -0.292512
stats_v3 <- analysis %>% group_by(disease) %>%
summarise(mean = mean(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva),
sd = sd(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva),
Q50 = quantile(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = 0.5),
Q25 = quantile(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = 0.25),
Q75 = quantile(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = 0.75),
min = min(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva),
max = max(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva),
FC = quantile(HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = 0.5)/Q50_all_v3,
n = n())
stats_v3 <- stats_v3 %>% mutate(
color = case_when(
Q50 <= Q25_all_v3 ~ "0_low",
Q50 >= Q75_all_v3 ~ "2_high",
Q50 > Q25_all_v3 & Q50 < Q75_all_v3 ~ "1_even"
)
)
analysis <- analysis %>% mutate(
color = c(rep(NA, nrow(analysis)))
)
## color matching
color_matching <- match(analysis$disease, stats_v3$disease)
analysis$color <- stats_v3$color[color_matching]
table(analysis$color)
## If log10 scale is better for y-axis, use "scale_y_log10() +" before "theme()"
ggplot(analysis, aes(x = disease, y = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, fill = color)) +
geom_violin(width = 1.2, trim = FALSE) +
geom_hline(yintercept = quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "pink")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/","HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva","_v3", ".tiff", sep = "")
tiff(filename = path,
width = 1200, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
ggplot(analysis, aes(x = disease, y = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, fill = color)) +
geom_violin(width = 1.2, trim = FALSE) +
geom_hline(yintercept = quantile(analysis$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "pink")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
dev.off()これで、高いもの低いもの、全体の値の範囲などが目に入ってくる図になったように思う。こうやって見るとなかなか新鮮な気がする。
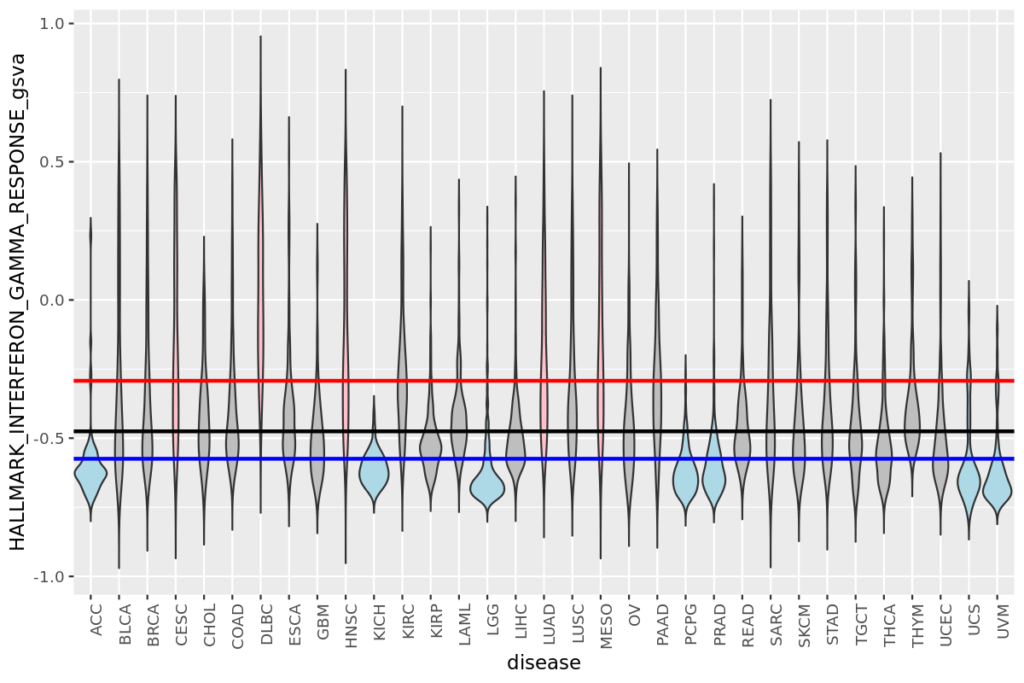
バージョン4;全遺伝子、全遺伝子セットのエンリッチメントスコアのバージョン3を計算し、どの遺伝子名や遺伝子セット名でもバージョン3のバイオリンプロットを表示出来るようにする
上記では確かにコントラストが付いて見やすいバイオリンプロットになったと思うが、しかしながら、着目する遺伝子や遺伝子セットを変えるたびに一々、HALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaとかCSNK2A1とか入れるのだろうか。正直、それは面倒くさい。じゃあ、HALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaとかCSNK2A1と一回入れるだけで済むようにしたら良い。
そのために少ないアタマで考えた結果、症例、疾患、遺伝子もしくは遺伝子セット等必要な列からなるデータフレームを、遺伝子もしくは遺伝子セット毎にリストに全部入れてしまって、表示したい遺伝子もしくは遺伝子セットを入力することで、それが表示出来るようにすれば良い。以下が行なった手順である。
遺伝子の発現量もしくは遺伝子セットのエンリッチメントスコアを、遺伝子もしくは遺伝子セット毎にリストにいれる
まずはデータフレームanalysisから、必要な列を取ってきて、それを遺伝子もしくは遺伝子セット毎にリストに入れていく。列1、列59490、列59491、列2から59424までは、それぞれ、列case、列disease_individual_order、列disease、各遺伝子発現量が入った列である。その値からデータフレームをリストgeneに入れていく。かなり大きなリストが出来上がるはず。そして、これを同様に各遺伝子セットのエンリッチメントスコア毎にリストに入れてき、リストenrichmentを作成する。そしてそのリストをc()で結合し、リストviolinを作成する。
遺伝子もしくは遺伝子セット毎に中央値、25パーセンタイル、75パーセンタイルを計算する
リストviolinの各要素には、症例毎の各遺伝子もしくは各遺伝子セットの発現量が入っているので、それらを使って、全症例の中央値、25パーセンタイル、75パーセンタイルを計算し、計算する。それぞれリストQ50_all、Q25_all、Q75_allに入れていき、names()でそれらの各要素に遺伝子名もしくは遺伝子セットの名前を付ける。
疾患毎に中央値、25パーセンタイル、75パーセンタイルを計算する
ここは、もともと少ない脳みそでけっこう悩んだところである。最初に一時的なデータフレームdfと一時的な値genesetを作る。リストviolinのi番目のデータをデータフレームdf入れ、値genesetには、そのデータフレームdfの名前、すなわち、遺伝子名もしくは遺伝子セット名を入れる。
次に、その一時的なデータフレームdfの列disease(BRCAとかLUADとか)の疾患名毎にgroup_by()で分類し、疾患毎に平均値(mean)、標準偏差(sd_each)、中央値(Q50_each)、25パーセンタイル(Q25_each)、75パーセンタイル(Q75_each)、最小値(min_each)、最大値(max_each)、全体の中央値に対する各疾患の中央値の比(FC_each)、各疾患の症例数(n_each)を計算する。このとき、dplyrパッケージ(調べてみるとrlangパッケージにもあるらしい)の.data[[]]という参照方法を用いた。これでデータフレームdfの中の列名がgenesetの列をダイナミックに参照することが出来る。マニュアルには、代名詞って書いてある。グローバルなデータフレームdfのうちの何とかという列を具体的に参照するのではなく、今回のコードのようにオブジェクトとして保存した文字列などから間接的に参照するっていう感じだろうか。今回のコードのようにfor()とかで参照する列がどんどん変わる場合は有効だった。実際、df[,4]ではエラーで計算出来なかったはずである。バージョン3と同じように各疾患の中央値(Q50_each)が、事前に計算した全体の25パーセンタイル以下だったり75パーセンタイル以上だったりする疾患に列colorとして後に色を付けるための値0_low、1_even、2_highを振った。これで全体の中央値にたいして、各疾患の中央値が高いのか低いのか同じなのか示す値を振ることが出来た。そして、つぎにmatch()を使って、症例毎にその色を振っていく。これをfor()でリスト全部に当てていく。これで、全遺伝子、全遺伝子セットでバージョン3と同じようなデータフレーム(実際には遺伝子もしくは遺伝子セット毎にリストに入っている)が出来上がる。
バージョン4のバイオリンプロットを表示する
言うても、どの遺伝子セットに着目しようか悩むところではあるので、遺伝子名や遺伝子セット名だけを入れたデータフレームgene_geneset_nameを作り、それを見ながら、興味のある遺伝子や遺伝セットを選ぶことにする。ここではバージョン1から3でも見たHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaにする。リストviolinからHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaとう要素を拾ってきてデータフレームgene_geneset_dfに入れ、それをまた[[]]を使って特定の列を参照する。リストviolinから拾ってきたデータフレームgene_geneset_dfのうち、オブジェクトgene_geneset_showに入った文字列の列(この例ではgene_geneset_dfの4列目の列名、すわなち文字列「HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva」)を参照できる。これらを使って、バージョン3と同じバイオリンプロットを出力する。
# code 51
# violin plot verson 4
### prepare data
# column 1; case
# column 59490; disease_individual_order
# column 59491; disease
# column 59470; project_id
# column 59699; Day
# column 59700; survival
# column 2:59423; gene expression
# column 59494:59697; results of enrichment
#### each gene
gene <- list()
for(i in 1:ncol(analysis[,2:59423])){
gene[[i]] <- analysis[,c(1, 59490, 59491, 2+i-1)]
}
names(gene) <- colnames(analysis[,2:59423])
#### each geneset
enrichment <- list()
for(i in 1:ncol(analysis[,59494:59697])){
enrichment[[i]] <- analysis[,c(1, 59490, 59491, 59494+i-1)]
}
names(enrichment) <- colnames(analysis[,59494:59697])
violin <- c(gene, enrichment)
#### stats for all disease
Q50_all <- list()
Q25_all <- list()
Q75_all <- list()
for(i in 1:length(violin)){
Q50_all[i] <- median(violin[[i]][,4])
Q25_all[[i]] <- quantile(violin[[i]][,4], probs = 0.25)
Q75_all[[i]] <- quantile(violin[[i]][,4], probs = 0.75)
}
names(Q50_all) <- names(violin)
names(Q25_all) <- names(violin)
names(Q75_all) <- names(violin)
#### stats for each signature
stats_v4 <- list()
for (i in 1:length(violin)) {
df <- violin[[i]]
geneset <- names(df)[4]
##### .data[[geneset]] is dynamic reference in data.frame and tibble, not list.
stats_v4[[i]] <- df %>% group_by(disease) %>%
summarise(
mean = mean(.data[[geneset]], na.rm = TRUE),
sd_each = sd(.data[[geneset]], na.rm = TRUE),
Q50_each = quantile(.data[[geneset]], probs = 0.5, na.rm = TRUE),
Q25_each = quantile(.data[[geneset]], probs = 0.25, na.rm = TRUE),
Q75_each = quantile(.data[[geneset]], probs = 0.75, na.rm = TRUE),
min_each = min(.data[[geneset]], na.rm = TRUE),
max_each = max(.data[[geneset]], na.rm = TRUE),
FC_each = quantile(.data[[geneset]], probs = 0.5, na.rm = TRUE) / Q50_all[[i]],
n_each = n(),
.groups = "drop"
) %>% mutate(
color = case_when(
Q50_each <= Q25_all[[i]] ~ "0_low",
Q50_each >= Q75_all[[i]] ~ "2_high",
Q50_each > Q25_all[[i]] & Q50_each < Q75_all[[i]] ~ "1_even"))
}
names(stats_v4) <- names(violin)
view(stats_v4[[1]])
#### color matching
color_matching <- list()
for (i in 1:length(violin)) {
color_matching[[i]] <- match(violin[[i]]$disease, stats_v4[[i]]$disease)
violin[[i]]$color <- stats_v4[[i]]$color[color_matching[[i]]]
}
table(violin[[3]]$color) # Check the code was running normally
#### If log10 scale is better for y-axis, use "scale_y_log10() +" before "theme()"
#### Check the gene or geneset name you need.
gene_geneset_name <- data.frame(target_gene_geneset = names(violin))
### -------------- run below to show violin plot v.4----------------
### Run following code to show violin plot of the gene or geneset of interest.
#### Specify gene or geneset to be shown.
gene_geneset_df <- violin[["HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva"]] #HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva #CSNK2A1
gene_geneset_show <- names(gene_geneset_df)[4]
#### show violin plot
ggplot(gene_geneset_df, aes(x = disease, y = .data[[gene_geneset_show]], fill = color)) +
geom_violin(width = 1.2, trim = FALSE) +
geom_hline(yintercept = quantile(gene_geneset_df[[gene_geneset_show]], probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "pink")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/",gene_geneset_show,"_v4", ".tiff", sep = "")
tiff(filename = path,
width = 1200, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
ggplot(gene_geneset_df, aes(x = disease, y = .data[[gene_geneset_show]], fill = color)) +
geom_violin(width = 1.2, trim = FALSE) +
geom_hline(yintercept = quantile(gene_geneset_df[[gene_geneset_show]], probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "pink")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
dev.off()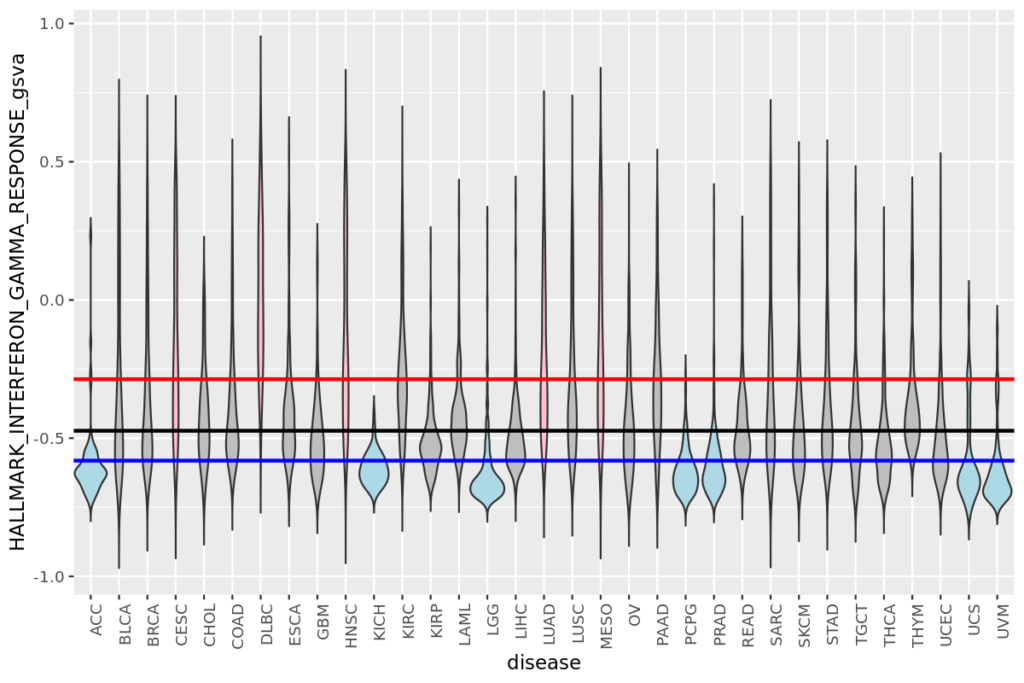
カプランマイヤー法による生存率の解析
ここから疾患毎の生存率をカプランマイヤー法で解析していく。
打ち切りの設定とデータフレームの準備
データフレームanalysisを使う。最初に、列vital_statusに入っている値をtable()で確認する。そうすると、値としてAlive、Dead、Not Reportedが入っているようだ。Not Reported以外は使えそうなので、AliveとDeadだけをfilter()で抽出し、新しくデータフレームanalysis_2を作成する。データフレームanalysis_2analysisを眺めていると、色々と使えそうな値が見つかった。列vital_status以外にも列days_to_death、列days_to_last_follow_upが使えそうなので、これらの値を使って打ち切りを当てていく。
次に、各疾患の生存が確認された期間を整理する。以下の表のように、列vital_status、列days_to_death、列days_to_last_follow_upと複数の値の組み合わせにより患者の生存期間を選んだ。ここを空欄にしてカプランマイヤーを計算するのももったいない。これにはif(){}、else_if(){}、else(){}を使った。事前に列days_to_last_follow_up_2にNAを必要な行数だけ準備し、それをas.numeric()で数値型にしておけば問題ないと思う。
| 条件文 | 列vital_status | 列days_to_death | 列days_to_last_follow_up | 新しく作る列days_to_last_follow_up_2に入れる値 |
|---|---|---|---|---|
| if() | Alive | NA | days_to_last_follow_upの値 | |
| else_if() | Dead | NA | days_to_deathの値 | |
| else_if() | Dead | NA | days_to_last_follow_upの値 | |
| else() | days_to_last_follow_upの値 |
これが出来たら、後の計算用に列days_to_last_follow_up_2の列名をわかりやすくDayに変えておいた。これがカプランマイヤー法による生存曲線を表示するときの横軸になる。ここでは、教科書(生存時間解析、ISBN978-4-254-12861-1、5ページ、表1.1)に従って打ち切り(Censor)を設定し、生存率を解析する。
TCGAのデータは、非常に長い期間フォローアップがされている症例を含んでいる。おそらく観察期間としては5年や、長くても10年くらいが適切なのではないだろうかと思う。なのでここでは観察期間を5年(365日x5年=1825日)に制限して、打ち切り(Censor)を設定するのもありだと思う。本当ならば、打ち切りを目的に依って変える必要があると思う。例えば、患者が生存していても、再発が検出されたり、フォローアップ期間中にCTを行ったら治療開始時よりも腫瘍体積が増加していた(Progressive Disease)ときに打ち切りにしたりする必要がある。これは解析の目的に応じて設定しなければならない。ここでは、列vital_statusがAliveかつDayが1825日以上である場合を列survivalを0(継続)、列vital_statusがDeadかつDayが1825日以上であれば列survivalを0(継続。後に死んだけど、5年は生存したから。)、vital_statusがAliveかつDayが1825日より前である場合を列survivalを1(フォローアップが何らかの理由により打ち切り)、列vital_statusがDeadかつDayが1825日より前であれば列survivalを1(死亡により打ち切り)として、カプランマイヤー法を計算するのもありなはず、というか、こちらの方が適切かもしれん。
# code 52
# check case with dead or alive.
table(analysis$vital_status)
# Alive Dead Not Reported
# 7108 2901 8
# case with "Not reported" will be removed.
analysis_2 <- analysis %>% filter(vital_status == "Alive" | vital_status == "Dead")
# prepare empty column.
analysis_2$days_to_last_follow_up_2 <- c(rep(NA, nrow(analysis_2))) %>% as.numeric()
for(i in 1:nrow(analysis_2)){
if(analysis_2$vital_status[i] == "Alive" & is.na(analysis_2$days_to_death[i]) == TRUE){
analysis_2$days_to_last_follow_up_2[i] <- analysis_2$days_to_last_follow_up[i]}
else if(analysis_2$vital_status[i] == "Dead"& is.na(analysis_2$days_to_last_follow_up[i]) == TRUE){
analysis_2$days_to_last_follow_up_2[i] <- analysis_2$days_to_death[i]}
else if(analysis_2$vital_status[i] == "Dead"& is.na(analysis_2$days_to_death[i]) == TRUE){
analysis_2$days_to_last_follow_up_2[i] <- analysis_2$days_to_last_follow_up[i]}
else{analysis_2$days_to_last_follow_up_2[i] <- analysis_2$days_to_last_follow_up[i]}
}
analysis_2 <- analysis_2 %>% filter(days_to_last_follow_up_2 > 0)
analysis_2 <- analysis_2 %>% rename("Day" = "days_to_last_follow_up_2")
analysis_2 <- analysis_2 %>% mutate(
survival = case_when(
vital_status == "Alive" & Day >= 1825 ~ 0,
vital_status == "Alive" & Day < 1825 ~ 1,
vital_status == "Dead" & Day >= 1825 ~ 0,
vital_status == "Dead" & Day < 1825 ~ 1,
)
)作成したデータフレームanalysis_2には、カプランマイヤー法の計算と曲線の表示に必要な列を抽出する。必要な列は、列1(case;case1とか)、列59490(disease_individual_order;ACC1とか)、列59491(disease;ACCとか)、列59470(project_id;TCGA-ACCとか)、 列59699(Day;上記で整えた値)、列 59700(survival;打ち切り)、 列2から59423(遺伝子発現量)、列 59494から59697(遺伝子セットのエンリッチメントスコア)である。これらを抽出してデータセットanalysis_3を作成する。遺伝子発現量や遺伝子セットのエンリッチメントスコアは症例の階層化に使用する。
生存率曲線を図示するにあたって、バイオリンプロットのバージョン4のように、こちらの興味のある遺伝子名や遺伝子セットを入力すれば、その遺伝子の発現量や遺伝子セットのエンリッチメントスコアの値によって全疾患に渡って表示され、互いに比較出来るようにしたい。そのためには、遺伝子もしくは遺伝子セット毎にリストに入れてしまうのが良いと思う。データフレームanalysis_3の1から6列目までは共通の値、7列目以降は遺伝子や遺伝子セットなので、これ毎にリストanalysis_survivalに入れればよい。リストanalysis_survivalには対応する遺伝子名もしくは遺伝子セット名を振っておく。
# code 53
# In addition to "case", "disease_individual_order", "disease", "project_id", Day","survival", choose required columns.
colnames(analysis_2[,59424:59493])
analysis_3 <- analysis_2[,c(1, 59490, 59491, 59470, 59699, 59700, 2:59423, 59494:59697)]
# column 1; case
# column 59490; disease_individual_order
# column 59491; disease
# column 59470; project_id
# column 59699; Day
# column 59700; survival
# column 2:59423; gene expression
# column 59494:59697; results of enrichment
analysis_survival <- list()
# view(analysis_3[,c(1:6,7)]) # check them
for(i in 7:ncol(analysis_3)){analysis_survival[[i-6]] <- analysis_3[,c(1:6,i)] %>% data.frame()}
names(analysis_survival) <- colnames(analysis_3[,7:ncol(analysis_3)])
次に患者を遺伝子発現量や遺伝子セットのエンリッチメントスコアの値で階層化する。先ず、遺伝子もしくは遺伝子セット名を値gene_name_survivalに入れる。後でこの名前が付いたデータフレームを参照するためにこのベクトルgene_name_survivalを使用する。次に、上記で作成したリストanalysis_survivalの各要素(各要素には症例、疾患、症例毎の生存期間、打ち切りの値、遺伝子発現量もしくは遺伝セットのエンリッチメントスコアが入っている)をデータフレームtemp_survival_dfに入れる。それを使って遺伝子もしくは遺伝子セットの中央値、25パーセンタイル、最初に中央値、25パーセンタイル、75パーセンタイル、症例数をgroup_by(disease)を使って疾患毎に計算し、それらをデータフレームtemp_survival_quantileに入れ、その値を改めてデータフレームtemp_survival_dfに入れる。データフレームtemp_survival_quantileには、疾患(33行しかない)毎にしか別れていないので、それをmatch()を使って、その疾患の症例全部に当てていく。次に出来上がったデータフレームtemp_survival_dfに対しmutate()とcase_when()を使って、遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアが、その疾患の中央値以上ならば列layer_2にhighを、中央値より低ければ列layer_2にlowを、25パーセンタイル以上ならば列layer_3にlowを、25パーセンタイルより高く75パーセンタイルより低ければ列layer_3にmidを、75パーセンタイル以上ならば列layer_3にhighを入れ、患者を2層と3層に階層化する。この列layer_2はhigh、lowの順で、列layer_3はhigh、mid、lowの順にfactor()で順序を付けておいた。これをやらないと、図毎に曲線の色が変わってしまって大変になる可能性がある。これを、for()を使ってリストanalysis_survivalの各要素で行う。これでカプランマイヤーの生存率解析をする準備が整った。
# code 54
# USE for(), DO NOT USE foreach(). foreach() spent huge amount of memory, which was more than 1 TB, and it took very long time to store the data for the execution.
for(i in 1:length(analysis_survival)){
# IMPORTANT; Need to use ".data[[]]" not "temp_survival_df"
gene_name_survival <- names(analysis_survival[[i]][7])
temp_survival_df <- analysis_survival[[i]] %>% data.frame()
temp_survival_quantile <- temp_survival_df %>% group_by(disease) %>%
summarise(Q50 = quantile(.data[[gene_name_survival]], probs = 0.50, na.rm = TRUE),
Q25 = quantile(.data[[gene_name_survival]], probs = 0.25, na.rm = TRUE),
Q75 = quantile(.data[[gene_name_survival]], probs = 0.75, na.rm = TRUE),
n = n())
## color matching
# color_matching <- match(analysis$disease, stats_EMT$disease)
# analysis$color <- stats_EMT$color[color_matching]
disease_matching <- match(temp_survival_df$disease, temp_survival_quantile$disease)
temp_survival_df$Q50 <- temp_survival_quantile$Q50[disease_matching]
temp_survival_df$Q25 <- temp_survival_quantile$Q25[disease_matching]
temp_survival_df$Q75 <- temp_survival_quantile$Q75[disease_matching]
analysis_survival[[i]] <- analysis_survival[[i]] %>%
mutate(
Q25 = temp_survival_df$Q25,
Q50 = temp_survival_df$Q50,
Q75 = temp_survival_df$Q75,
layer_2 = case_when(analysis_survival[[i]][[gene_name_survival]] >= temp_survival_df$Q50 ~ "high",
analysis_survival[[i]][[gene_name_survival]] < temp_survival_df$Q50 ~ "low"),
layer_3 = case_when(analysis_survival[[i]][[gene_name_survival]] >= temp_survival_df$Q75 ~ "high",
analysis_survival[[i]][[gene_name_survival]] > temp_survival_df$Q25 & analysis_survival[[i]][[gene_name_survival]] < temp_survival_df$Q75 ~ "mid",
analysis_survival[[i]][[gene_name_survival]] <= temp_survival_df$Q25 ~ "low")
)
# set order for layer_2
analysis_survival[[i]]$layer_2 <-
factor(analysis_survival[[i]]$layer_2,
levels = c("high", "low"),
labels = c("high", "low"),
exclude = NA, ordered = is.ordered(analysis_survival[[i]]$layer_2), nmax = NA)
# set order for layer_3
analysis_survival[[i]]$layer_3 <-
factor(analysis_survival[[i]]$layer_3,
levels = c("high", "mid", "low"),
labels = c("high", "mid", "low"),
exclude = NA, ordered = is.ordered(analysis_survival[[i]]$layer_3), nmax = NA)
}
# check data
view(analysis_survival[[2]])
table(analysis_survival[[1]]$layer_2)
table(analysis_survival[[1]]$layer_3)生存曲線を図示する
ここで、バイオリンプロットのバージョン4の場合と同様に、どの遺伝子もしくは遺伝子セットに着目するかを見る必要がある。そのためには、遺伝子名もしくは遺伝子セット名を拾って来てデータフレームgene_name_survival_2を作成し、それを眺めれば良い。気になる遺伝子もしくは遺伝子セットが見つかったら、それを値gene_geneset_interestに入れれば良い。ここでは、バイオリンプロットでもみたHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaについて着目して解析しようと思う。
念のために、疾患名が33個あるかどうか、データフレームdisease_nameを作って確認している。これは、リストanalysis_survivalのどの要素を入れても、33個の疾患が抽出されてくるはずである。
# code 55
gene_name_survival_2 <- data.frame(gene_geneset = names(analysis_survival)) # for looking up gene or geneset of interest
view(gene_name_survival_2) # look for gene or geneset of interest
gene_geneset_interest <- "HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva" # put gene or geneset of interest for survival curve
# HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva
# CSNK2A1
#
disease_name <- analysis_survival[[1]]$disease %>% unique() %>% data.frame() # any number is OK.
colnames(disease_name) = "disease"この解析の目的としては、着目する遺伝子もしくは遺伝子セットが、どの疾患の患者生存率に統計的有意な影響を与えるかを比較することである。なので、疾患毎に「一気に」比較したい。なので、疾患毎にリストanalysis_survivalから、着目する遺伝子もしくは遺伝子セットのデータフレーム、ここではHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaのデータフレームを取ってきて、それをさらにリストinterest_gene_geneset_survival_1に疾患毎に入れていく。本当は33個の疾患を一気に図示したいのだが、正直、33個の図を一つの画面で表示するのはなかなか無理がある(33個のパネルが一つの図に載っている論文も見たこと無いし。せいぜいfig. 2j、fig. 3mとか、10個とか13個とかまでである。)ので、最初は1から18個の疾患を表示する。
生存率の解析にはsurvivalパッケージのSurv()とsurvfit()を、生存率の表示にはsurvminerパッケージのggsurvplot()を使う。このsurvminerパッケージはいろいろな手法を実装しているので、必要だったら試してみるのは良いと思う。
Surv()でイベント(ここではDeadかAliveか)が起こった時間(ここではDay)を拾って来て、それをsurvfit()に渡して、生存期間の中央値や95%信頼区間を推定する。それをここではfor()を使って18後の疾患に当てていき、その結果をリストfit_status_1に入れて行く。そして、リストfit_status_1の各要素毎にggsurvplot()により生存曲線を作成していく。このggsurvplot()はログランク検定で、ここで設定した階層間の生存期間に統計的有意な差があるのかを検定してくれる。
上述のように、この解析の目的としては各疾患毎の生存率を互いに比較することなので、この18個の生存率をパネルにして一つの図上で表示する。そのためにgrid.arrange()を活用した。ggsurvplot()の出力結果を入れたリストsurvival_curve_status_1の各要素は、着目した遺伝子もしくは遺伝子セット(ここではHALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)の各疾患の生存率の情報がリストになって入っている。そのオブジェクトのうちのplotというリストには、図の情報が入っているので、それを各疾患毎に取り出してきてリストsurvival_curve_status_1_graphに入れる。そして、それをgrid.arrange()を使って、パネルにする。ここでは6列のパネルにして表示した。
# code 56
# 1 - 18 project.
# prepare list
interest_gene_geneset_survival_1 <- list()
for(i in 1:18){
interest_gene_geneset_survival_1[[i]] <- analysis_survival[[gene_geneset_interest[1]]] %>% filter(disease == disease_name$disease[i])
} # If length(disease_name) was used, the all 33 project were put in the list. 18 means only 18 projects were put in the list.
names(interest_gene_geneset_survival_1) <- disease_name$disease[1:18]
fit_status_1 <- list() # 1 - 15
survival_curve_status_1 <- list() # 1 - 15
length(interest_gene_geneset_survival_1) # 18 diseases
for(i in 1:length(interest_gene_geneset_survival_1)){
fit_status_1[[i]] <- survfit(Surv(Day, survival) ~ layer_2, data = interest_gene_geneset_survival_1[[i]])
survival_curve_status_1[[i]] <- ggsurvplot(
fit_status_1[[i]],
data = interest_gene_geneset_survival_1[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6),
font.legend = c(6),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i])
} # legend = c("right", "left", "top", "bottom", "none") can show which color is which layer.
# red; high
# green; low
survival_curve_status_1_graph <- lapply(survival_curve_status_1, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_1_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/",gene_geneset_interest,"_2_layer_1",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_1_graph, ncol = 6)
dev.off()以下の図が出力である。良ければtiff()を使って出力しておく。
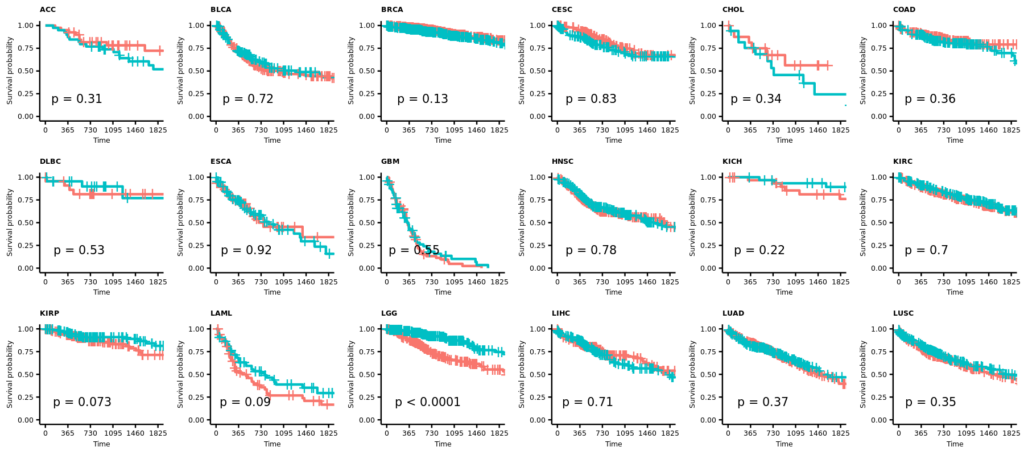
続いて同じように残りの疾患についても同様の方法で生存曲線を表示する。
# code 57
# 19 - 33 project.
# prepare list
interest_gene_geneset_survival_2 <- list()
for(i in 19:33){
interest_gene_geneset_survival_2[[i-18]] <- analysis_survival[[gene_geneset_interest[1]]] %>% filter(disease == disease_name$disease[i])
} # If length(disease_name) was used, the all 33 project were put in the list. 18 means only 18 projects were put in the list.
names(interest_gene_geneset_survival_2) <- disease_name$disease[19:33]
fit_status_2 <- list() # 1 - 15
survival_curve_status_2 <- list() # 1 - 15
length(interest_gene_geneset_survival_2) # 15 diseases
for(i in 1:length(interest_gene_geneset_survival_2)){
fit_status_2[[i]] <- survfit(Surv(Day, survival) ~ layer_2, data = interest_gene_geneset_survival_2[[i]])
survival_curve_status_2[[i]] <- ggsurvplot(
fit_status_2[[i]],
data = interest_gene_geneset_survival_2[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "bold", "black"),
font.y = c(6, "bold", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i+18])
} # legend = c("right", "left", "top", "bottom", "none") can show which color is which layer.
# red; high
# green; low
survival_curve_status_2_graph <- lapply(survival_curve_status_2, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_2_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/",gene_geneset_interest,"_2_layer_2",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_2_graph, ncol = 6)
dev.off()いかが残りの疾患である。ここでもgrid.arrange()で6列で出力しているが、残りは15個なので、3行目は3列しか出力されていない。これも大丈夫そうならばtiff()で保存する。
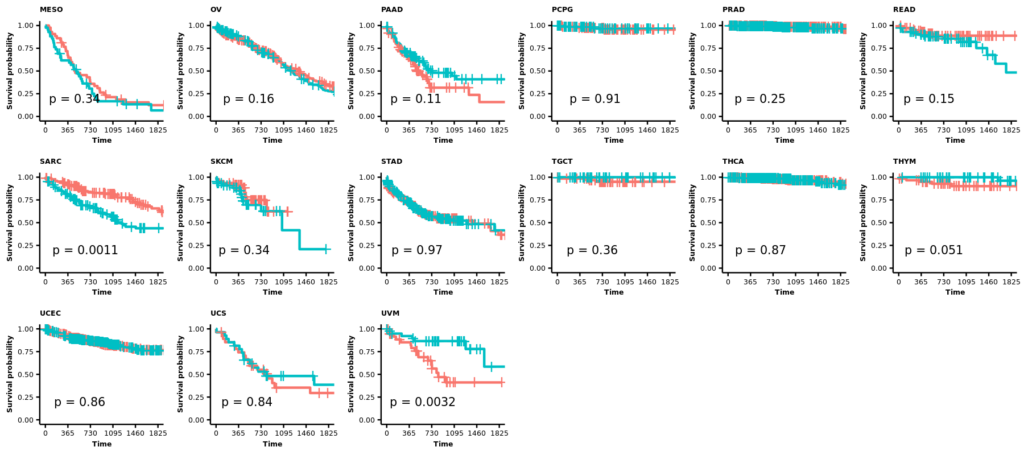
続いて、列layer_3、すなわち、階層化をhigh、mid、lowの3層での生存率の違いを解析する場合である。この場合のp値は、どの群とどの群に違いがあるのかはわからない。この3層でなんか統計的有意な違いがあることだけを示している。ここで、highとlowに明確な差がありそうなことが示された、次の解析でこのmidで分類される症例を除いてしまい、改めて検定してみれば良い。むしろ、そちらの方が標的遺伝子や標的となる疾患の同定には有用だろうと思う。
以下が、列layer_3での階層化の場合で、疾患1から18までである。
# code 58
# 1 - 18 project.
fit_status_3 <- list() # 1 - 15
survival_curve_status_3 <- list() # 1 - 15
length(interest_gene_geneset_survival_1) # 18 diseases
for(i in 1:length(interest_gene_geneset_survival_1)){
fit_status_3[[i]] <- survfit(Surv(Day, survival) ~ layer_3, data = interest_gene_geneset_survival_1[[i]])
survival_curve_status_3[[i]] <- ggsurvplot(
fit_status_3[[i]],
data = interest_gene_geneset_survival_1[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i])
}# legend = "bottom") can show which color is which layer.
# red; high
# green; mid
# blue; low
survival_curve_status_3_graph <- lapply(survival_curve_status_3, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_3_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/",gene_geneset_interest,"_3_layer_1",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_3_graph, ncol = 6)
dev.off()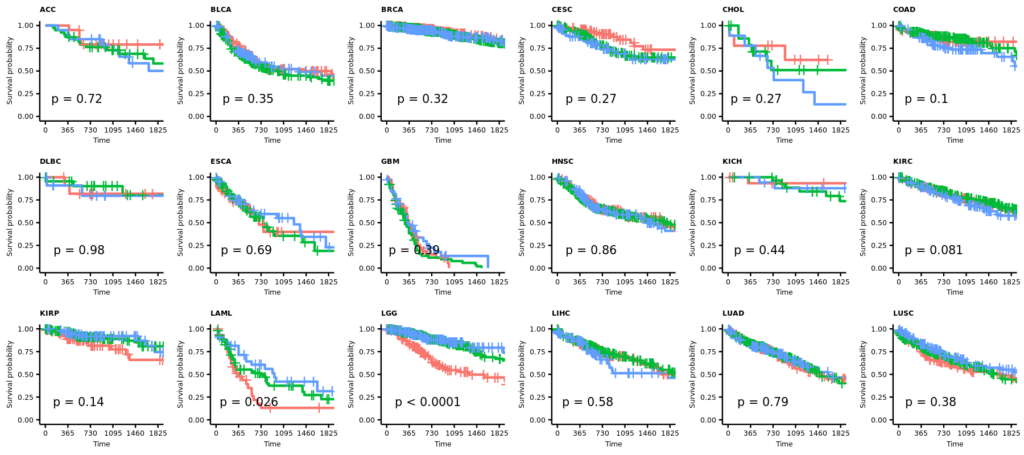
以下が、列layer_3での階層化の場合で、疾患19から33までである。
# code 59
# 19 - 33 project.
fit_status_4 <- list() # 1 - 15
survival_curve_status_4 <- list() # 1 - 15
length(interest_gene_geneset_survival_2) # 15 diseases
for(i in 1:length(interest_gene_geneset_survival_2)){
fit_status_4[[i]] <- survfit(Surv(Day, survival) ~ layer_3, data = interest_gene_geneset_survival_2[[i]])
survival_curve_status_4[[i]] <- ggsurvplot(
fit_status_4[[i]],
data = interest_gene_geneset_survival_2[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i+18])
} # legend = "bottom") can show which color is which layer.
# red; high
# green; mid
# blue; low
survival_curve_status_4_graph <- lapply(survival_curve_status_4, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_4_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/",gene_geneset_interest,"_3_layer_2",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_4_graph, ncol = 6)
dev.off()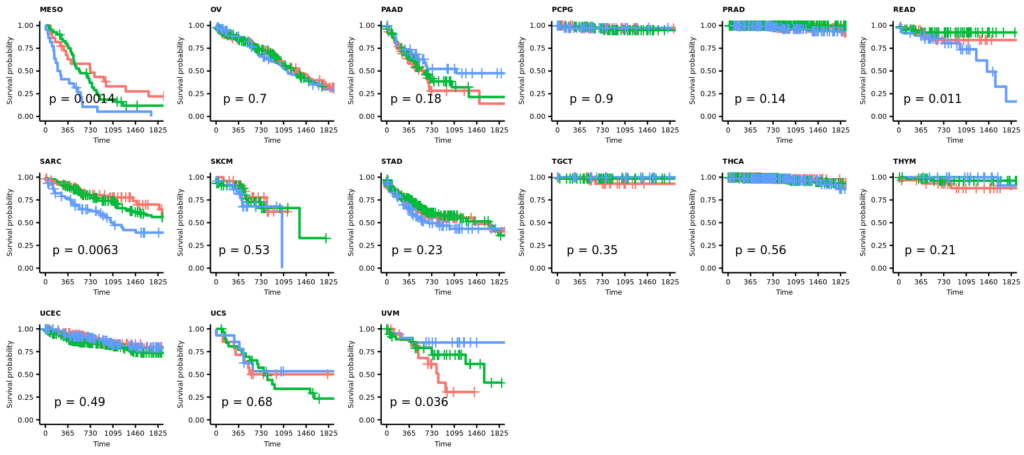
ログランク検定とその結果をまとめる
図示できたとは言え、これを全遺伝子及び全遺伝子セットで個々に見ていくのは非常に辛いし、現実的ではない。結局、一次スクリーニングのような値が必要である。個人的には、ベタにログランク検定のp値を使って選んでくるのが良いような気がする。なので、各遺伝子もしくは各遺伝子セットの各疾患毎に列layer_2と列Layer_3の階層化毎にロクランク検定を行い、リストにいれ、最後にreduce()で全部結合してデータフレームlogrank_pval_dfにして保存する。これで、ざっくりとどの遺伝子もしくはどの遺伝子セットがどの疾患の患者生存率に統計的有意な影響を与えそうか、ということを拾ってくることが出来る。
この計算の途中、ある遺伝子でこのログランク検定出来ない疾患が出てきた。何事だと思って確認したら、どうやらその遺伝子の全症例にわたる中央値が0で、その値と各疾患の遺伝子発現量を比較しても階層化できないということになっていたらしい。考えてみればこれはある意味バグであり、訂正しなければならない。これを論文などに落とし込むためには、ある遺伝子に関してある症例の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアが、その疾患に渡る中央値と等しい場合、何か別の層を入れる必要がありそうだ。
# code 60
# 2025 10 11
# prepare dataset
temp_logrank_1 <- list()
logrank <- list()
for(x in 1:length(analysis_survival)){
for(y in 1:length(disease_name$disease)){
temp_logrank_1[[y]] <- analysis_survival[[x]] %>% data.frame() %>% filter(disease == disease_name$disease[y])
}
names(temp_logrank_1) <- disease_name$disease
logrank[[x]] <- temp_logrank_1
}
names(logrank) <- names(analysis_survival)
# calculate log-rank for all disease over all gene and geneset
temp_dataframe <- data.frame(disease = disease_name$disease, layer_2_pval=rep(NA, 33), layer_3_pval=rep(NA, 33))
logrank_pval <- list()
for(x in 1:length(analysis_survival)){
for(y in 1:length(disease_name$disease)){
temp_df <- logrank[[x]][[y]] %>% data.frame()
temp_dataframe$layer_2_pval[y] <- tryCatch({survdiff(Surv(Day, survival) ~ layer_2, data = temp_df)$p}, error = function(e) {NA})
temp_dataframe$layer_3_pval[y] <- tryCatch({survdiff(Surv(Day, survival) ~ layer_3, data = temp_df)$p}, error = function(e) {NA})
}
logrank_pval[[x]] <- temp_dataframe
}
names(logrank_pval) <- names(analysis_survival)
# FDR correction and name the each column
for(i in 1:length(logrank_pval)){
logrank_pval[[i]] <- logrank_pval[[i]] %>% mutate(
layer_2_pval_FDR = tryCatch({p.adjust(layer_2_pval, method = "fdr")}, error = function(e) {NA}),
layer_3_pval_FDR = tryCatch({p.adjust(layer_3_pval, method = "fdr")}, error = function(e) {NA}),
gene_geneset = names(logrank_pval[i])
)
}
# get the list together to a data frame
logrank_pval_df <- reduce(logrank_pval, bind_rows) %>% data.frame()出来上がった全疾患の全遺伝子もしくは全遺伝子セットにわたるログランク検定の結果をまとめたデータフレームlogrank_pval_dfは、ずいぶんと大きくて見やすいとは言えないので、例えば以下のように、膵がん患者の生存期間に影響を与えそうな遺伝子や遺伝子セットは何か?みたいな感じで探すのが良いように思う。こうしてみると結構な数の遺伝子が生存期間に関わっていることがわかる。
# code 61
# example; which genes and geneset are closely associated the survival of PAAD patients??
PAAD_significant <- logrank_pval_df %>% filter(disease == "PAAD" & (layer_2_pval_FDR < 0.05))データセット全体を保存する
いつでも引っ張り出せるように、データセット全体を保存しておく。
# code 62
save.image("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/2024 11 02 TCGA survival.RData")考えてみれば、これらをshinyとかでダッシュボード的なものを作れば、いつでもパッと確認できるじゃあないか。そのために、重要と考えられる個々のリストやデータフレームも保存しておく。
# code 63
saveRDS(analysis_survival, "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/2024 11 02 TCGA survival.rds")
saveRDS(stats_v4, "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/2024 11 02 TCGA violin.rds")
saveRDS(histogram, "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/2024 11 02 TCGA histogram.rds")最後に、最終保存日とか、この解析に使ったパッケージ等をテキストで保存しておく。
# code 64
file.remove("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/save_log.txt")
file.remove("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/sessionInfo.txt")
cat(paste0("The analysis was saved at ", Sys.time(), "\n"), file = "/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/save_log.txt")
sink("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/sessionInfo.txt")
sessionInfo()
sink()
追加の解析
後になって気づいたのだが、この解析では正常組織の群を入れなかった。患者のメタデータには正常組織の情報も入っていたのだが、GDC portalから正常組織のRNA-seqのカウント値のデータをダウンロードしなかったので、解析出来なかった。これにはちょっと後悔している。薬の開発では、正常組織と腫瘍における標的遺伝子、タンパク質の発現量の差が議論になるから、せっかくだからダウンロードすれば良かったと思う。これは今後改めて解析してみようと思う。
また、これだけ患者数がたくさんあると、例えばシングルセルRNAシークエンスのように遺伝子発現を元に患者を分類して、疾患とサブタイプで分けて分析し、そのサブタイプが生存率に与える影響を解析したり出来る。
また、ここで作成したバイオリンプロットのバージョン4や生存率曲線はShinyでいつでも表示できるようにもしよと思う。誰かにこういった解析を求められたときに、まとめて提出できるようにしておくのも一興である。この解析では、今後のためにこのような「カタログ」作成を目的としているので、shinyに落とし込むのは良いと思う。
まとめ
以上がTCGAのデータセットを使った患者の生存率解析である。正直言えば、Rのパッケージを使うことでもっと簡単にデータを取得できるのだが、正直、いつアップデートされたのかわからんし、そのパッケージの使い方とか覚えるのもなんかダルいし、そのとき使ったパッケージが開発停止になったり、さらに良いパッケージが出てくる可能性があり、いちいちマニュアルを見る、みたいなことなり、「これで正しいのだろうか…」とか思ったときには結局元のTCGAのインストラクションを読み出すのがオチである。だったら、はじめから素性のしれた元データを解析した方が再現性は良いだろう、という考えでTCGAのデータを解析した。
この解析をやっていて思ったのが、Rはメモリの扱いが難しいということだった。Pythonに比べると計算もかなり遅いように思う。
2026/01/12(月)追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う
上記で、RでAnndataを使おうとすると結局Reticulateパッケージを使う羽目になってしまい、それだったらRでやるわ、という話を書いたが、そう言えば、Rでもここで取り扱っている遺伝子発現量に症例毎の診療情報やシークエンスに使用されたサンプルの情報を効率良く扱うことが出来るパッケージがあった。SummerizedExperimentである。これを使えば、Anndataのようなデータの構造で解析を行うことが出来ることに気がついた。SummerizedExperimentは、まさにAnndataのように行と列の順番が一致している必要が有るが、それをうまいことリンクさせてそのような順番に依らないようにしたフォーマットのMultipleAssayExperimentというパッケージもあるらしい。これらを上手に使うことで、メモリを節約しながら解析を進めることが出来るはず、と思っている。