2026/01/03(土);初稿
2026/01/12(月);追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う。
2026/06/13(土);記事が長すぎてCodecプラグインが動かなかったので、コードをSTORESで販売することにした。
- 1 はじめに
- 2 注意点1
- 3 注意点2
- 4 GDC portalからTCGAのカウント値をダウンロードする
- 5 ここからのRのダウンロード(文章を読みたくない人用)1
- 6 使用するパッケージ
- 7 RNA-seqのカウントデータを読み込む
- 8 カウントデータと臨床情報を関連付ける
- 9 Anndataオブジェクトを作成(Optional)
- 10 ここからのRのダウンロード(文章を読みたくない人用)2
- 11 escapeとGSVAを用いたエンリッチメント解析
- 12 ヒストグラムとバイオリンプロットによる疾患毎の遺伝子量またはエンリッチメントスコアの比較
- 13 カプランマイヤー法による生存率の解析
- 14 解析環境を記録しておく
- 15 まとめ
- 16 ここからのRのダウンロード(文章を読みたくない人用)3
- 17 2026/01/12(月)追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う
はじめに
以前、TCGAのデータを使って、がんや腫瘍における遺伝子発現量(RNA-seqのカウント値)と患者の生存率の関係をRで解析したが、そのときは対応する正常組織(おそらくがんや腫瘍周辺の正常部位)におけるRNA-seqのカウン値をダウンロードしなかったため、それらをバイオリンプロットを用いた疾患毎の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアの比較に含めることが出来なかった。それに、TCGAのデータも2024年のバージョン41であり、ちょっと時間が経っていたので、改めてダウンロードし、解析することにした。
早速本題に入ろうと思う。
注意点1
以前の解析との違いは、以下である。
- 正常組織の遺伝子発現量を解析、特にバイオリンプロットに含めた。
- GDC portalからTCGA(とCPTAC)で採られたRNA-seqのカウント値をダウンロードした。
- ssGSEA2とfgseaは計算が遅すぎたので使用しなかった。escapeとGSVAのみでGene Set Enrichment Analysis(GSEA)を行った。
- MSigDBに登録されている(2025年10月時点)ヒトの全遺伝子セットを使ってGSEAした。
ここでやっている解析は以前の解析とほぼ同じであり、もしかしたら以前の解析の方が良いかもしれない。特に、後ほどバイオリンプロットで各疾患毎さらに対応する正常組織の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアを比較するが、正直、結論を得るためには正常組織の症例数が足りないように思うし、当然ながら正常組織を採取できない症例や疾患もあるためだ。正常組織も解析に含めてみて、これに気がついた。今なら、「正常組織も解析に含めることは出来ますが、症例数がちょっと足りないようにも思いますし、そもそも正常組織を採っていない疾患も多いです。もしかしたら、正常組織との比較は参考程度にしかならず、それで何かを決定するのは微妙かもしれません。」と自信を持って言える。
なお、CPTACについては今後、CPTACで採られたタンパク質発現量と遺伝子発現量の相関を解析するためである。ここではデータのクリーニングのときにちょっとだけ出てくるが、この記事ではそれらを解析しない。以下に比較表を載せておく。
| 項目 | この記事 | 以前の記事 |
|---|---|---|
| タイトル | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.43, 2025) | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.41, 2024) |
| TCGAのバージョン | v.43, 2025 | v.41, 2024 |
| GDC portalからのデータのダウンロード方法 | あり | なし |
| GMTファイルの作り方 | なし | あり(ただし、Geneontologyから選んできた遺伝子セット1つだけ) |
| 使ったGMTファイル | MsigDBに登録されているヒトの遺伝子セット全部(Human MSigDB v2025.1.Hs、2025年10月時点) | Geneontologyから選んできて自分で作成したHRR(Homologous Recombination Repair)と、MsigDBのH: hallmark gene sets(Human MSigDB v2025.1.Hs、2025年10月時点)のみ。 |
| エンリッチメント解析 | GSVA escape | ssGSEA2 escape GVSA fgsea |
| バイオリンプロットによる疾患毎の遺伝子発現量もしくは遺伝子セットのエンリッチメントの比較 | 正常組織あり ウィルコクソンの順位和検定あり(ただし、正常組織の例数が腫瘍組織に比べて少ないのでちょっと注意が必要と思う。) | 常組織なし ウィルコクソンの順位和検定なし |
| 生存期間解析でのログランク検定 | あり | あり |
| その他 | 比較するデータの数(リストの要素の数)が多く、そのために少しデータの準備がすこし複雑でメモリを喰いまくる。 | 比較するデータの数(リストの要素の数)がそんなに多くないので、データの準備がそんなに大変じゃない。 |
注意点2
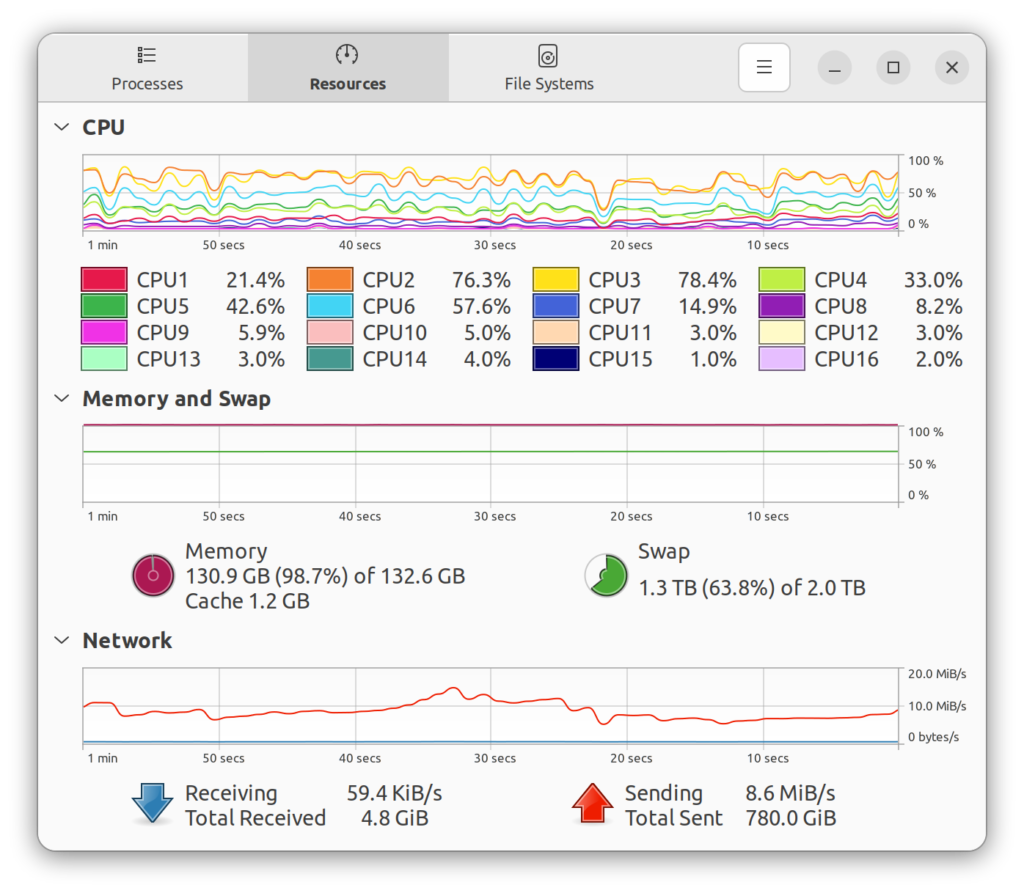
以前の解析でも書いたが、この解析で使用するコンピューターの性能はずいぶんと高いと思う。以下の画像の通りメモリを合計で1.3TB以上も使っており、たとえRandom Access Memory(RAM)(物理メモリ)を256GB積んでいたとしても全く足りていないことがわかる。ちなみに、これはデータセットの全部がすべて整って、生存率曲線の解析とそれに続くログランク検定を行っているとき(3つめのRのコード)のメモリの使用量である。1つ目から3つ目のコードを一つのRStudioに入れようとしても、作業領域が一杯になってまともに動かなくなってしまう。また、昨今のノートPCや軽量のラップトップでは計算能力が足りない可能性があるので、要注意である。もはやサーバーのようなコンピューターが必要と言える。
GDC portalからTCGAのカウント値をダウンロードする
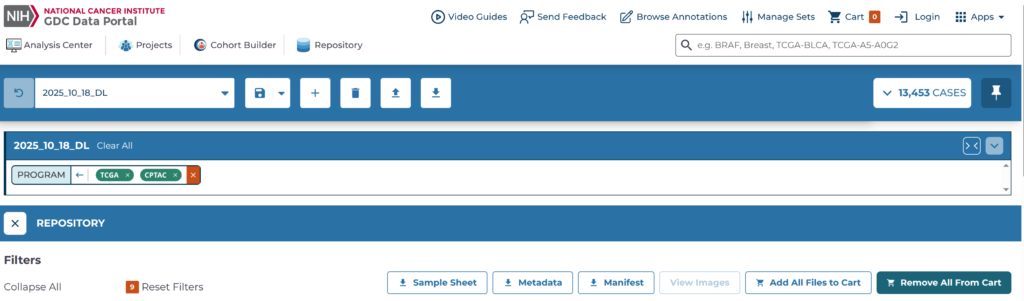
まずは、GDC portal(https://portal.gdc.cancer.gov/)からTCGAとCPTACのRNA-seqのカウント値をダウンロードする。ダウンロード方法については以前の記事が詳しいので、そちらを参照したほうが良いかもしれない。GDC portalは以下のようなフロントエンド(2025年12月時点)である。まず「Cohort Builder」を押す。
「Cohort Builder」を押すと、以下のようなページに入る。「Program」というパネルには様々なプロジェクト(の大元)があるので、TCGAとCPTACにチェックを入れて、そのコホートに名前を付けて保存する。こうすれば一々TCGAとCPTACを選び直して…ということが無くなる。ここでは2025_10_18_DLという名前で保存した。次に、「Repository」をクリックする。
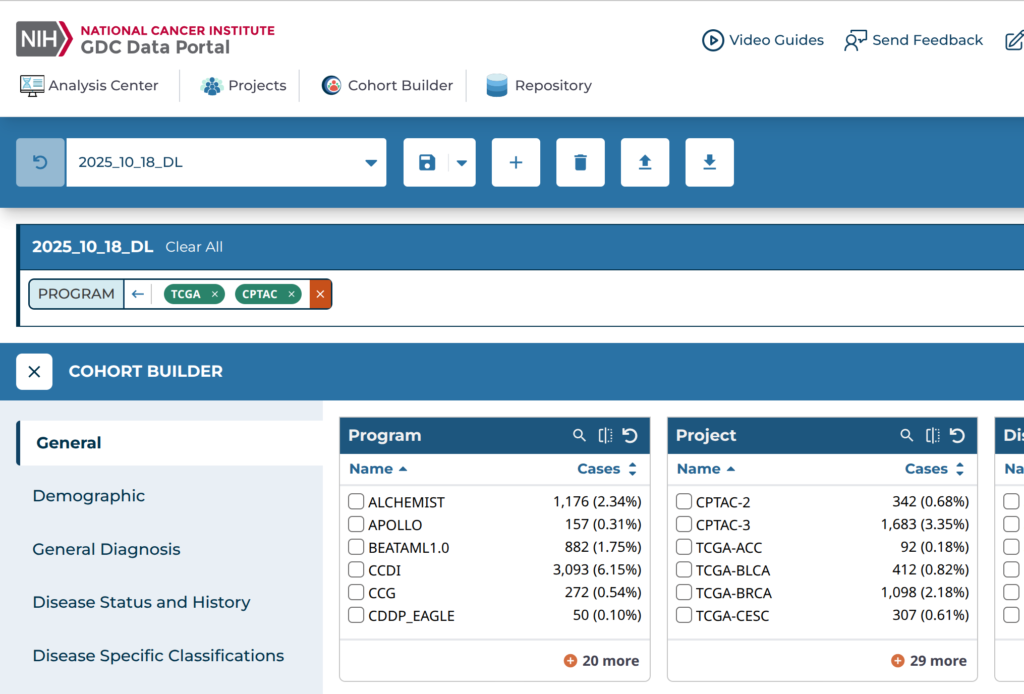
| Cohort Builder | 値 |
|---|---|
| Program | TCGA CPTAC |
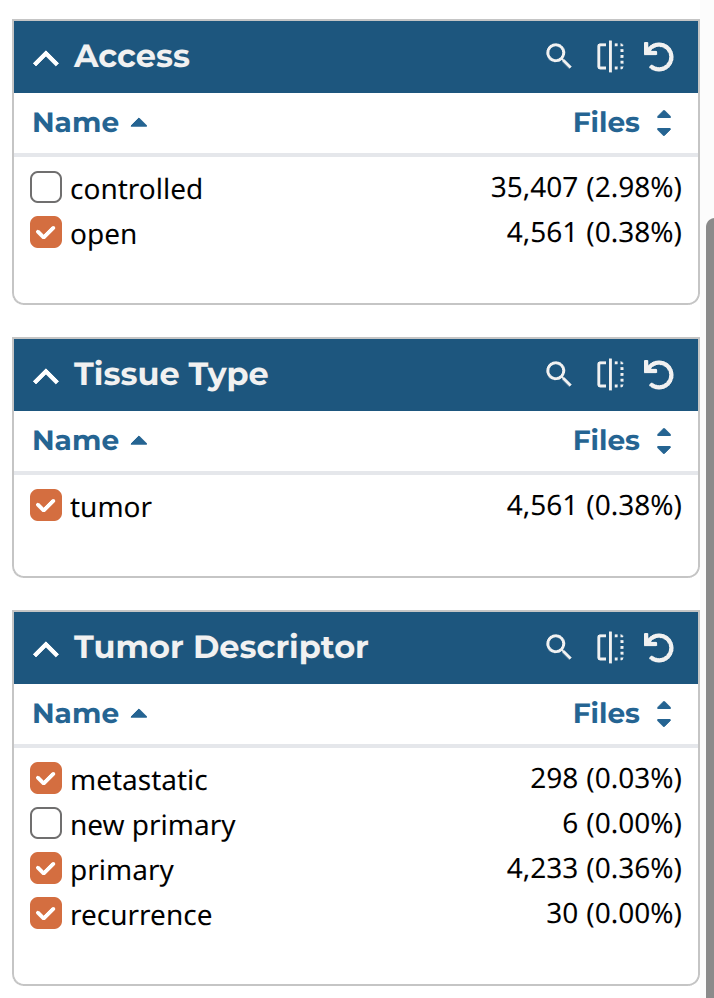
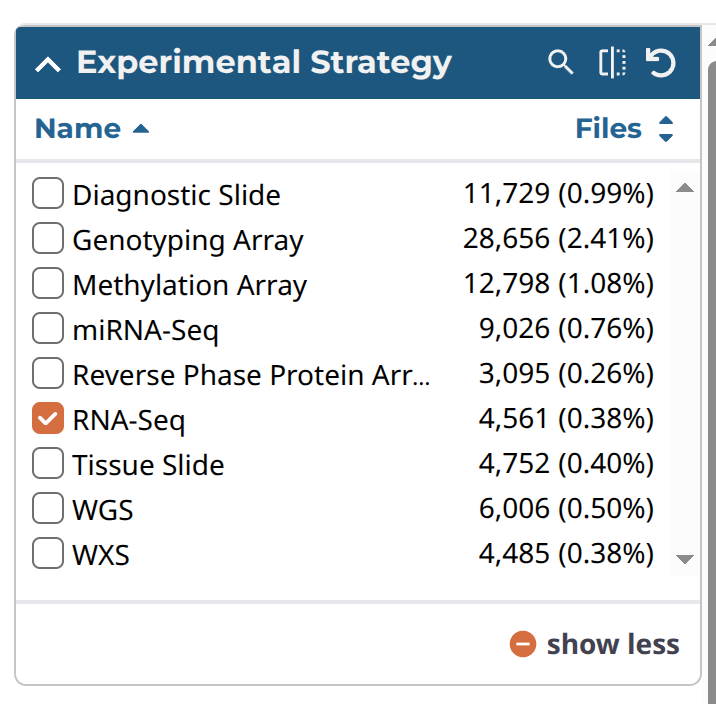
次に「Repository」をクリックし、Filterのパネルのところを、以下の表のようにチェックを入れていく。なお、ダウンロードしたい症例はCartに入れてからまとめてmanifest fileなどをダウンロードするが、このとき、Cartには10,000ファイルしか入らなかったと思うので、以下のように順番1から3のように分割して、Cartに入れるファイル数を10,000より少なくしてからダウンロードすることにした。
ダウンロード1回目
まずは1回目のダウンロードが以下の表である。
| Filter | 値 | 順番 |
|---|---|---|
| Access | open | 1 |
| Experimental Strategy | RNA-seq | 1 |
| Tissue type | tumor | 1 |
| Tissue Descriptor | metastatic primary recurrence | 1 |
| Preserved Method | ffpe frozen oct | 1 |
各パネルの画像は以下のような感じである。
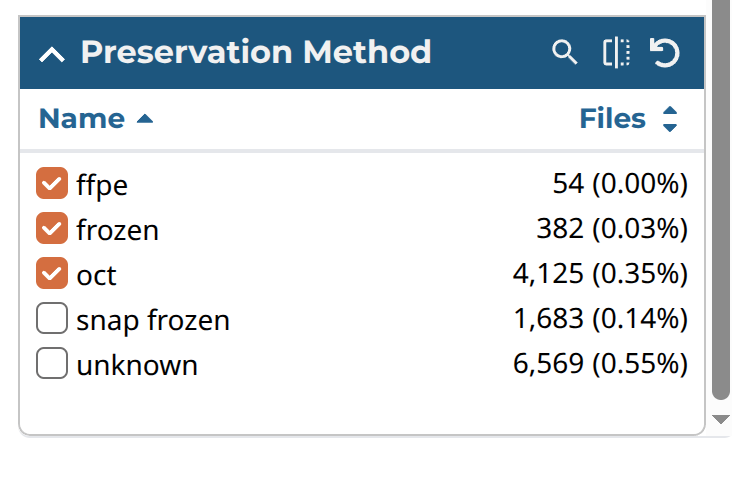

上記を選んだら、「Add All Files to Cart」をクリックし、次に右上にある「Cart」をクリックする。
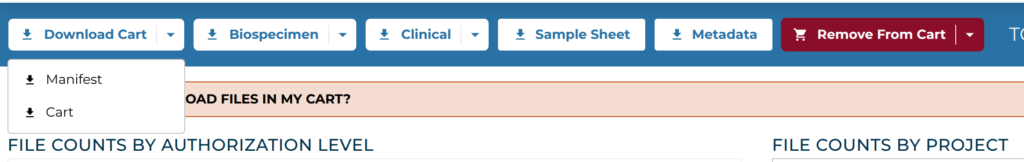
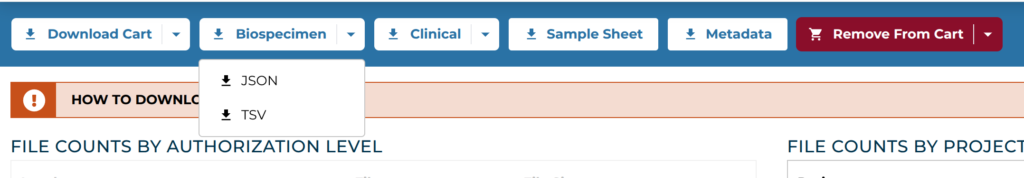
カートに移動したら、Manifestファイル、サンプルに関するデータ(Biospecimen)、臨床情報に関するデータ(Clinical)、Sample Sheet、Metadataをダウンロードする。Sample SheetとMetadataは表示の「Sample Sheet」、「Metadata」のところをクリックするだけである。他は以下のようにダウンロードする。BiospecimenとClinicalに関するファイルは圧縮されているので、適宜展開する。
ダウンロード2回目
以下がダウンロード2回目の値である。対象となるファイル選んでカートにいれたら、カートに移動して手順1の通り各種ファイルをダウンロードする。
| Filter | 値 | 順番 |
|---|---|---|
| Access | open | 2 |
| Experimental Strategy | RNA-seq | 2 |
| Tissue type | tumor | 2 |
| Tissue Descriptor | metastatic primary recurrence | 2 |
| Preserved Method | snap frozen unkown | 2 |
ダウンロード3回目
以下がダウンロード3回目の値である。3回目は正常組織のみを選んだ。これは、後ほどバイオリンプロットにより疾患間の遺伝子発現量もしくは遺伝子セットのエンリッチメントスコアを比較するときに、正常組織の発現量も加えるためである。手順1と2と同様に、対象となるファイル選んでカートにいれたら、カートに移動してダウンロード1回目の通り各種ファイルをダウンロードする。
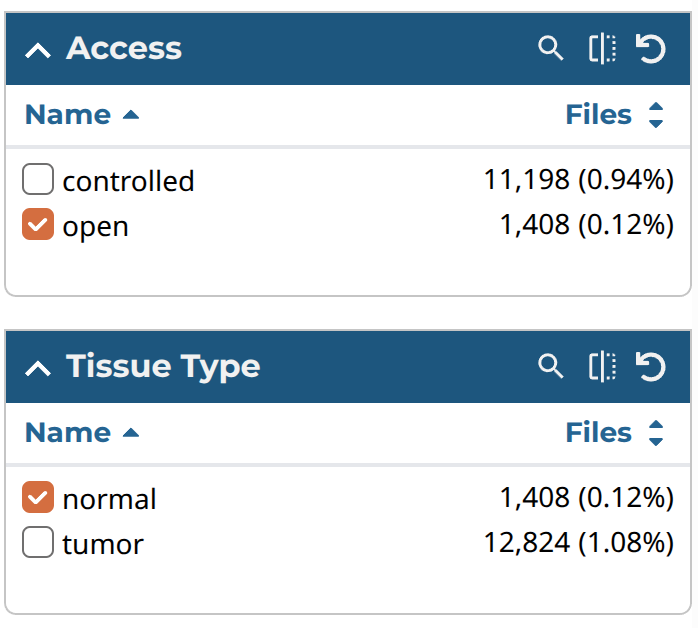
| Filter | 値 | 順番 |
|---|---|---|
| Access | open | 3 |
| Experimental Strategy | RNA-seq | 3 |
| Tissue type | normal | 3 |

gdc-clientを使ってManifest fileに記載されたファイルをダウンロードする
以下のコードがManifest fileを使ったRNA-seqのカウント値のダウンロード方法である。gdc-clientをここからダウンロードし、インストールする必要がある。これらの方法は以前の記事にある程度詳しく書いてある。以下がgdc-clientを使ったダウンロードの方法である。
# 1
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest.2025-10-25.154055.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest_1_log.txt"
# 2
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_manifest.2025-10-25.160033.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_manifest_2_log.txt"
# 3
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest.2025-10-25.160847.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest_3_log.txt"
# 3 retry
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest.2025-10-26.024514.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_manifest_3_retry_log.txt"
# 1 retry
gdc-client download -m "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest.2025-10-26.103837.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_manifest_1_retry_log.txt"
# 2 retry
/mnt/team4tb/Dropbox/Downloads Safari/gdc_manifest.2025-10-27.003414.txt
gdc-client download -m "/mnt/team4tb/Dropbox/Downloads Safari/gdc_manifest.2025-10-27.003414.txt" -d "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor" --log-file "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_manifest_2_retry_log.txt"
ここからのRのダウンロード(文章を読みたくない人用)1
使用するパッケージ
以下のパッケージが自分がいつもRNA-seqの解析に使っているものである。TCGAのデータを使った生存率解析のためのパッケージとして必須なものは、tidyverse、data.table、suvival、survminer、 grid.Extraである。それに加えて、この解析ではescapeとgsvaを用いる。
# DEG
library(baySeq)
library(edgeR)
library(DESeq2)
library(limma)
# enrichment analysis
library(clusterProfiler)
library(fgsea)
library(ssGSEA2) # devtools::install_github("nicolerg/ssGSEA2") # devtools::install_github(broadinstitute/ssGSEA2.0) and devtools::install_github(broadinstitute/ssGSEA2) did not work.
library(escape)
library(GSVA)
library(pathview)
# immune cell deconvolution
library(ConsensusTME)
library(estimate)
library(immunedeconv)
library(MCPcounter)
library(msigdbr)
library(quantiseqr)
library(TIMER) #devtools::install_github('hanfeisun/TIMER'); it will not work!! use TIMER of immunodeconv if you need.
library(xCell)
# stats
library(NSM3) # sudo apt install libgmp-dev libgmp10 libgmp3-dev
library(pwr)
library(missForest)
# basic
library(beeswarm)
library(circlize)
library(org.Hs.eg.db)
library(org.Mm.eg.db)
library(doParallel)
library(foreach)
library(parallel) # Need it for use of baySeq
library(pheatmap)
library(rtracklayer)
library(rvest) # this is for extraction of html table; results of leading edge analysis.
library(xml2) # this is for extraction of html table; results of leading edge analysis.
library(openxlsx) # for read xlsx file.
library(ComplexHeatmap)
library(tidyverse)
library(data.table)
library(survminer)
library(survival)
library(gridExtra)
library(MASS)
library(coin)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(RhpcBLASctl)
# # shiny
# library(shiny)
# library(bslib)
# Check global environment
Sys.getenv()RNA-seqのカウントデータを読み込む
list.files()を使ってGDC portalからダウンロードしたファイルのパスを全部抽出し、それをベクトルall_file_tumorに入れる。そこからファイルの末尾がcounts.tsvで終わっているファイル、つまり、RNA-seqのカウント値のTSVファイルだけを選んできて、それをベクトルfile_path_tumorに入れる。これを正常組織のTSVファイルに対しても行いfile_path_normalを作成する。
# code 1
# tumor
all_file_tumor <- list.files("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor", all.files = TRUE, full.names = TRUE, recursive = TRUE)
file_path_tumor <- all_file_tumor[str_detect(pattern = "counts.tsv$", string = all_file_tumor)]
# normal
all_file_normal <- list.files("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal", all.files = TRUE, full.names = TRUE, recursive = TRUE)
file_path_normal <- all_file_normal[str_detect(pattern = "counts.tsv$", string = all_file_normal)]ファイルパスが入ったベクトルfile_path_tumorとfile_path_normalから、.tsvより前の文字列、すなわちファイル名だけを抜き出して、データフレームfile_name_tumorとfile_name_normalを作る。
# code 2
file_name_tumor <- sub("^.*/", "" , file_path_tumor) %>% data.frame()
file_name_normal <- sub("^.*/", "" , file_path_normal) %>% data.frame()code 1で、ファイルパスが入ったベクトルfile_path_tumorとfile_path_normalを作ったので、これを使って実際にファイルを読み込んでいき、それをリストcount_tumorに入れる。このファイルのうち、必要なのは1列目のensembl gene ID、2列目の遺伝子名、7列目のTPM正規化された遺伝子発現量が必要なので、それ以外は除いてしまう。読み込んだら、それをreduce()を使ってinner_join()し、データフレームcount_merge_tumorを作成する。それをwrite_tsv()でTCGA_primary_count_merge_tumor.tsvとして保存する。正常組織のファイルも同様に処理し、データフレームcount_merge_normalを作成し、write_tsv()でTCGA_primary_count_merge_normal.tsvとして保存しておく。
# code 3
# tumor
count_tumor<- list()
for (i in 1:length(file_path_tumor)) {
count_tumor[[i]] <- read.table(
file = file_path_tumor[i],
sep = "\t",
header = TRUE)
count_tumor[[i]] <- count_tumor[[i]][-c(1,2,3,4),]
count_tumor[[i]] <- count_tumor[[i]][,c(1, 2, 7)]
colnames(count_tumor[[i]]) <- c("gene_id","gene_name", file_name_tumor[i,])
} # This takes so long time if you are using HDD.
count_merge_tumor <- purrr::reduce(count_tumor, inner_join)
write_tsv(count_merge_tumor, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/TCGA_primary_count_merge_tumor.tsv")
gc()
gc()
# normal
count_normal<- list()
for (i in 1:length(file_path_normal)) {
count_normal[[i]] <- read.table(
file = file_path_normal[i],
sep = "\t",
header = TRUE)
count_normal[[i]] <- count_normal[[i]][-c(1,2,3,4),]
count_normal[[i]] <- count_normal[[i]][,c(1, 2, 7)]
colnames(count_normal[[i]]) <- c("gene_id","gene_name", file_name_normal[i,])
} # This takes so long time if you are using HDD.
count_merge_normal <- purrr::reduce(count_normal, inner_join)
write_tsv(count_merge_normal, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/TCGA_primary_count_merge_normal.tsv")
gc()
gc()念のために、読み込んだファイルに重複がないかどうか確認しておく。データフレームcount_merge_tumorの列名はファイル名になっているはずなので、それをデータフレームcheck_columns_tumorとして作成する。また同様に正常組織でもデータフレームcheck_columns_normalを作成する。そして、%in%を使って同じファイル名が無いことを確認しておく。%in%はmatch()と同じである。
# code 4
check_columns_tumor <- colnames(count_merge_tumor) %>% data.frame() %>% rename("tumor" = ".")
check_columns_normal <- colnames(count_merge_normal) %>% data.frame() %>% rename("normal" = ".")
overlapping_files <- check_columns_tumor$tumor %in% check_columns_normal$normal %>% data.frame() %>% rename("overlapping_files" = ".")
table(overlapping_files$overlapping_files)ファイルの読み込みに関してはこれで終了である。sessionInfo()で使用したパッケージのバージョンを確認しておく。
# code 5
# 2025 09 20
# save.image("/mnt/seqdata/public_data/Blog/TCGA/output/primary_analyte_merged.tsv")
# load("/mnt/seqdata/public_data/Blog/TCGA/output/primary_analyte_merged.tsv")
sessionInfo()カウントデータと臨床情報を関連付ける
code 3で腫瘍と正常組織のRNA-seqカウントデータを結合させ、出力しておいたので、それを改めて読み込む。fread()を使ってそれらを読み込む。ここでの注意点としてはdata.table = FALSEとすることである。そうしないと、データフレームではなくテーブル(表)として読み込まれてしまい、後の処理に支障が出てくる。
# code 6
# tumor
# Read each files. column name is for each case.
original_count_tumor <- fread(
input = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/TCGA_primary_count_merge_tumor.tsv",
sep = "\t",
encoding = "UTF-8",
data.table = FALSE)
# normal
# Read each files. column name is for each case.
original_count_normal <- fread(
input = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/TCGA_primary_count_merge_normal.tsv",
sep = "\t",
encoding = "UTF-8",
data.table = FALSE)後になってファイル名が重複していると非常に邪魔くさいので、いちいち重複を確認する。
データフレームoriginal_count_tumorの3列目から後ろの列名、すなわち、カウント値のファイル名をcolnames()で取ってきて、それを列名がcase_idというデータフレームcase_tumorを作成する。これらの列名に重複があるかどうかは。group_by()で列case_idをグループ化して、それが1つ以上ある物をfilter()で抽出し、あたらしくデータフレームcheck_case_tumor_Duplicatedを作る。もし重複がなければ、値が何も入っていないデータフレームが出来上がる。ファイル名の重複は基本的にないはず。もし在ったら、このファイル名の症例をGDC portalなり、後に扱う症例毎の臨床情報を確認した方が良い。これを正常組織でも行う。重複がなさそうならば、データフレームoriginal_count_tumorとoriginal_count_normalを、列gene_idと列gene_nameを軸にしてinner_join()する。これで、各疾患毎のカウント値の大本のデータフレームoriginal_countを作成できた。気が付いたのだが、出来上がったデータフレームoriginal_countでも、というか、むしろこちらの列名、すなわちファイル名の重複を確認しなければならない。でも、どうやらやっていないらしい。
# code 7
# tumor
## Extract each case and check if dataset contains duplicated case.
case_tumor <- colnames(original_count_tumor)[3:ncol(original_count_tumor)] %>% data.frame() %>% rename("case_id" = ".")
check_case_tumor_Duplicated <- case_tumor %>% group_by(case_id) %>% filter(n() > 1) %>% nrow() # 0 case. There is no duplicated case
# normal
## Extract each case and check if dataset contains duplicated case.
case_normal <- colnames(original_count_normal)[3:ncol(original_count_tumor)] %>% data.frame() %>% rename("case_id" = ".")
check_case_tumor_Duplicated <- case_tumor %>% group_by(case_id) %>% filter(n() > 1) %>% nrow() # 0 case. There is no duplicated case
colnames(original_count_tumor) %in% colnames(original_count_normal) %>% table() # gene_id and gene_name are same.
# FALSE TRUE
# 12813 2
# these TRUE 2 cases are gene_id and gene_name.
# merge original_count_tumor and original_count_normal by gene_id and gene_name
original_count <- inner_join(original_count_tumor, original_count_normal, by=c("gene_id", "gene_name"))
症例毎の診療情報や検体の情報を読み込む。ここは、GDCportalからカウント値のデータとは別にダウンロードしたファイルである。これらはTSVファイルで入手できるので、それをそのまま読んで、最後にreduce()を使って各TSVファイルをrbind()すれば良い。これらはtidyverseパッケージを使ってクリーニングしていくので、一応、tibble型にしておく。
# code 8
# tumor
## Read all metadata
aliquot_tumor <- list()
analyte_tumor <- list()
clinical_tumor <- list()
exposure_tumor <- list()
family_history_tumor <- list()
follow_up_tumor <- list()
pathology_detail_tumor <- list()
portion_tumor <- list()
sample_tumor <- list()
slide_tumor <- list()
samplesheet_tumor <- list()
aliquot_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/aliquot.tsv")
aliquot_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/aliquot.tsv")
aliquot_tumor_merged <- purrr::reduce(aliquot_tumor, rbind)
analyte_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/analyte.tsv")
analyte_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/analyte.tsv")
analyte_tumor_merged <- purrr::reduce(analyte_tumor, rbind)
clinical_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/clinical.tsv")
clinical_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/clinical.tsv")
clinical_tumor_merged <- purrr::reduce(clinical_tumor, rbind)
exposure_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/exposure.tsv")
exposure_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/exposure.tsv")
exposure_tumor_merged <- purrr::reduce(exposure_tumor, rbind)
family_history_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/family_history.tsv")
family_history_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/family_history.tsv")
family_history_tumor_merged <- purrr::reduce(family_history_tumor, rbind)
follow_up_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/follow_up.tsv")
follow_up_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/follow_up.tsv")
follow_up_tumor_merged <- purrr::reduce(follow_up_tumor, rbind)
pathology_detail_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/pathology_detail.tsv")
pathology_detail_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/pathology_detail.tsv")
pathology_detail_tumor_merged <- purrr::reduce(pathology_detail_tumor, rbind)
portion_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/portion.tsv")
portion_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/portion.tsv")
portion_tumor_merged <- purrr::reduce(portion_tumor, rbind)
sample_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/sample.tsv")
sample_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/sample.tsv")
sample_tumor_merged <- purrr::reduce(sample_tumor, rbind)
slide_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/slide.tsv")
slide_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/slide.tsv")
slide_tumor_merged <- purrr::reduce(slide_tumor, rbind)
samplesheet_tumor[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_sample_sheet.2025-10-25.tsv")
samplesheet_tumor[[2]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_sample_sheet.2025-10-25 (1).tsv")
samplesheet_tumor_merged <- purrr::reduce(samplesheet_tumor, rbind)
write_tsv(aliquot_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/aliquot_tumor_merged.tsv")
write_tsv(analyte_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/analyte_tumor_merged.tsv")
write_tsv(clinical_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/clinical_tumor_merged.tsv")
write_tsv(exposure_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/exposure_tumor_merged.tsv")
write_tsv(family_history_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/family_history_tumor_merged.tsv")
write_tsv(follow_up_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/follow_up_tumor_merged.tsv")
write_tsv(pathology_detail_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/pathology_detail_tumor_merged.tsv")
write_tsv(portion_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/portion_tumor_merged.tsv")
write_tsv(sample_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/sample_tumor_merged.tsv")
write_tsv(slide_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/slide_tumor_merged.tsv")
write_tsv(samplesheet_tumor_merged, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/samplesheet_tumor_merged.tsv")
# normal
## Read all metadata
aliquot_normal <- list()
analyte_normal <- list()
clinical_normal <- list()
exposure_normal <- list()
family_history_normal <- list()
follow_up_normal <- list()
pathology_detail_normal <- list()
portion_normal <- list()
sample_normal <- list()
slide_normal <- list()
samplesheet_normal <- list()
aliquot_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/aliquot.tsv")
analyte_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/analyte.tsv")
clinical_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/clinical.tsv")
exposure_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/exposure.tsv")
family_history_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/family_history.tsv")
follow_up_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/follow_up.tsv")
pathology_detail_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/pathology_detail.tsv")
portion_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/portion.tsv")
sample_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/sample.tsv")
slide_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/slide.tsv")
samplesheet_normal[[1]] <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_sample_sheet.2025-10-25.tsv")
write_tsv(aliquot_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/aliquot_normal_merged.tsv")
write_tsv(analyte_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/analyte_normal_merged.tsv")
write_tsv(clinical_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/clinical_normal_merged.tsv")
write_tsv(exposure_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/exposure_normal_merged.tsv")
write_tsv(family_history_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/family_history_normal_merged.tsv")
write_tsv(follow_up_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/follow_up_normal_merged.tsv")
write_tsv(pathology_detail_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/pathology_detail_normal_merged.tsv")
write_tsv(portion_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/portion_normal_merged.tsv")
write_tsv(sample_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/sample_normal_merged.tsv")
write_tsv(slide_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/slide_normal_merged.tsv")
write_tsv(samplesheet_normal[[1]], "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/samplesheet_normal_merged.tsv")
aliquot_normal <- aliquot_normal[[1]] %>% as_tibble()
analyte_normal <- analyte_normal[[1]] %>% as_tibble()
clinical_normal <- clinical_normal[[1]] %>% as_tibble()
exposure_normal <- exposure_normal[[1]] %>% as_tibble()
family_history_normal <- family_history_normal[[1]] %>% as_tibble()
follow_up_normal <- follow_up_normal[[1]] %>% as_tibble()
pathology_detail_normal <- pathology_detail_normal[[1]] %>% as_tibble()
portion_normal <- portion_normal[[1]] %>% as_tibble()
sample_normal <- sample_normal[[1]] %>% as_tibble()
slide_normal <- slide_normal[[1]] %>% as_tibble()
samplesheet_normal <- samplesheet_normal[[1]] %>% as_tibble()
最初に作製した患者情報のデータフレームのうち、データフレームsamplesheet_tumor_mergedとsamplesheet_normalの列名を直す。列名に空欄とかが入っていると、参照しにくくなる。なので、列名にある空欄をアンダーバーに置き換える。
# code 9
# Change column name of samplesheet for later sataset preparation.
samplesheet_tumor_merged <- samplesheet_tumor_merged %>% rename(
"file_id" = "File ID",
"file_name" = "File Name",
"data_category" = "Data Category",
"data_type" = "Data Type",
"project_id" = "Project ID",
"case_id" = "Case ID",
"sample_id"="Sample ID",
"tissue_type" = "Tissue Type",
"tumor_discription" = "Tumor Descriptor",
"speciemen_type" = "Specimen Type",
"preservation_method" = "Preservation Method")
samplesheet_normal <- samplesheet_normal %>% rename(
"file_id" = "File ID",
"file_name" = "File Name",
"data_category" = "Data Category",
"data_type" = "Data Type",
"project_id" = "Project ID",
"case_id" = "Case ID",
"sample_id"="Sample ID",
"tissue_type" = "Tissue Type",
"tumor_discription" = "Tumor Descriptor",
"speciemen_type" = "Specimen Type",
"preservation_method" = "Preservation Method")
colnames(samplesheet_normal)データフレームsamplesheet_tumor_mergedの列file_id、列file_nameが、データフレームsamplesheet_normalのそれらと重複していないか確認する。
# code 10
ncol(samplesheet_tumor_merged)
nrow(samplesheet_tumor_merged)
ncol(samplesheet_normal)
nrow(samplesheet_normal)
samplesheet_tumor_merged$file_id %in% samplesheet_normal$file_id %>% table() # All false
samplesheet_tumor_merged$file_name %in% samplesheet_normal$file_name %>% table() # All false
samplesheet_merged <- rbind(samplesheet_tumor_merged, samplesheet_normal)
ncol(samplesheet_merged)
nrow(samplesheet_merged) # 12813 + 1408データフレームclinical_tumor_mergedとclinical_normalをrbind()で結合し、データフレームclinical_mergedを作成する。
# code 11
ncol(clinical_tumor_merged)
nrow(clinical_tumor_merged)
ncol(clinical_normal)
nrow(clinical_normal)
clinical_merged <- rbind(clinical_tumor_merged, clinical_normal)
ncol(clinical_merged)
nrow(clinical_merged)データフレームclinical_mergedには、欠損値として「’–」という謎すぎる値が入っている。これをRでの欠損値NAに置き換える。
ここで、最悪なことに気が付く。なんと列名に接頭辞として意味の分からん分類、例えばcasesとかdemographicとかdiagnosesとかtreatmentsなどが付いており、それが「.」で区切られているじゃあないか。2024年のデータセットにはなかったのに…というか、TCGAはなんでこんな余計なもん付けたんだろう。こんな変な分類、どこにもリンクしてないんだから付けるだけむだだろうが。しかも、なんで寄りによって「.」なんかを区切り文字にしたんだろう。Rなら大丈夫だが、pythonだとだいぶと混乱するきがするのだが(メソッドと一緒だし)。なんだこのセンスのない列名は。言うててもショウガいないので、この「.」をアンダーバーに置き換える。そして、後に解析に必要そうな列をデータフレームrequired_values_for_surivivalとしてまとめて、その列をselect(all_of())で取得し、データフレームclinical_merged_required_survivalを作成する。
気にならないなら、列名にドットが入ったままでも一向に構わない。ただし、なんとなくどこかで不都合が出てくるような気がする。こういう変な文字は列の途中に入れない方が良いと思うのだが….
# code 12
# Clean up clinical.tsv
# Replace "'--" to "NA".
clinical_merged[clinical_merged == "'--"] <- NA
colnames(clinical_merged)
# rename all columns. A kind of dot "." was inserted in each columns compared with a dataset at 2024. This dot is some kind of identifier to distingish which colum is which category, but I think this dot makes us very confusing. If the python is used for the analysis, it very similar to the mothod of library/package. At 2025, TCGA is not crever. Therefore, the dot should be replaced into "_".
column_name_clinical_merged <- colnames(clinical_merged) %>% data.frame() %>% rename("column" = ".")
column_name_clinical_merged_2 <- gsub("\\.", "_", column_name_clinical_merged$column)
view(column_name_clinical_merged_2)
colnames(clinical_merged) <- column_name_clinical_merged_2
# Clinical information must be include survival period. Therefore, the dataset must be filtered by that survival data is true.
required_values_for_surivival <- c(
"cases_case_id",
"cases_submitter_id",
"project_project_id",
"demographic_days_to_death",
"diagnoses_days_to_last_follow_up",
"diagnoses_age_at_diagnosis",
"demographic_days_to_birth",
"demographic_vital_status",
"demographic_year_of_birth",
"demographic_year_of_death",
"diagnoses_year_of_diagnosis",
"treatments_days_to_treatment_end",
"treatments_days_to_treatment_end",
"treatments_treatment_outcome",
"treatments_treatment_outcome_duration",
"treatments_treatment_type",
"diagnoses_primary_diagnosis",
"diagnoses_site_of_resection_or_biopsy",
"diagnoses_tissue_or_organ_of_origin",
"diagnoses_diagnosis_is_primary_disease",
"diagnoses_days_to_last_known_disease_status",
"diagnoses_days_to_recurrence",
"diagnoses_classification_of_tumor")
clinical_merged_required_survival <- clinical_merged %>% select(all_of(required_values_for_surivival))
作成したデータフレームから、列demographic_days_to_deathもしくは列diagnoses_days_to_last_follow_upがNAの症例を除く。これらは解析の最後のカプランマイヤーによる生存率解析が出来ないので、ここで除いてしまう。これらを除いたところで、症例数としては十分と考えている。
# code 13
clinical_merged_required_survival_2 <- clinical_merged_required_survival %>%
filter(demographic_days_to_death != "NA" | diagnoses_days_to_last_follow_up != "NA")
症例毎の診療情報が保持されたデータフレームclinical_merged_required_survival_2において、重複している症例に関して確認をする。列cases_case_idをgroup_by()でグループ化し、n()でその頻度をカウントし、それが1より多い例をfilter()で拾ってくる。そして、その数を確認すると、なんと45456例もある。列cases_submitter_idでも同じようにカウントしてみたところ、やっぱり45456例ある。なんてこった。
# code 14
# "case_id" holds the data like "004d251-3f70-4395-b175-c94c2f5b1b81".
# extract the record with duplicated "case_id."
check_case_id <- clinical_merged_required_survival_2 %>% group_by(cases_case_id) %>% filter(n() > 1)
nrow(check_case_id) # 45456 cases
# "case_submitter_id" holds data like "TCGA-DD-AAVP".
# extract the record with duplicated "case_submitter_id".
check_case_submitter_id <- clinical_merged_required_survival_2 %>% group_by(cases_submitter_id) %>% filter(n() > 1)
nrow(check_case_submitter_id) # 45456 casesどうやら、これらの重複例は同一症例であり、例えば、過去にRadiotherapyを受けたり、Chemotherapyを受けたり、とそれ毎にレコードが作成されているようだ。フォローアップの日数など生存率の解析に必要な値はどうやら同じようなので、distinct()で重複例を除いてしまう。いきなり作成する前に、distinct()により何例残るのか、一時的にデータフレームcheck_case_id_distinctやcheck_case_submitter_id_distinctを作って確認した。どうやら重複除去後は11679例になるらしい。
# code 15
check_case_id_distinct <- clinical_merged_required_survival_2 %>% distinct(cases_case_id, .keep_all = TRUE)
nrow(check_case_id_distinct) # 11679 rows
check_case_submitter_id_distinct <- clinical_merged_required_survival_2 %>% distinct(cases_submitter_id, .keep_all = TRUE)
nrow(check_case_submitter_id_distinct) # 11679 rowsdistinct()により重複を機械的に除いてしまっても大丈夫そうだったので、重複除去後の臨床情報データフレームclinical_merged_required_survival_de_duplicatedを作成する。
# code 16
clinical_merged_required_survival_de_duplicated <- clinical_merged_required_survival_2 %>% distinct(cases_case_id, .keep_all = TRUE)
nrow(clinical_merged_required_survival_de_duplicated) # 11679 rowsデータフレームclinical_merged_required_survival_de_duplicatedには、どんな疾患が登録されているか確認する。そして、後に有用そうだったのが列diagnoses_primary_diagnosisだった。この列の値をtable()で表にし、それをデータフレームdf_primary_diagnosisとして作成する。これをwrite_tsv()でdf_diagnosis.tsvというファイル名で保存する。
# code 17
# Check what kind of tumor type is there.
table(clinical_merged_required_survival_de_duplicated$diagnoses_classification_of_tumor)
# I think all values of "classification_of_tumor" is about primary tumor. For example, if the value is "metastasis", it means that metastatic tumor was found as "primary tumor". Therefore, there is no need to filter the data with classification_of_tumor.
# These data.frames are for check what kind of cancer type is registered??
df_primary_diagnosis <- table(clinical_merged_required_survival_de_duplicated$diagnoses_primary_diagnosis) %>% data.frame()
df_tissue_or_organ_of_origin <- table(clinical_merged_required_survival_de_duplicated$diagnoses_tissue_or_organ_of_origin) %>% data.frame()
df_diagnosis_x_tissue_or_organ_of_origin <- table(clinical_merged_required_survival_de_duplicated$diagnoses_primary_diagnosis, clinical_merged_required_survival_de_duplicated$diagnoses_tissue_or_organ_of_origin) %>% data.frame()
write_tsv(df_primary_diagnosis, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/df_diagnosis.tsv")
データフレームsamplesheetの情報をクリーニングする。このsamplesheetには、そのサンプルがどの組織から採られたのか(列samples.days_to_collection)、病理で確認されたのか(列samples.diagnosis_pathollogically_comfirmed)、FFPEなのか(列samples.is_ffpe)など、そのデータの元になった組織に関する情報、すなわちサンプル情報が載っているデータフレームのようだ。特に、列cases.case_idなんかは、カウント値、臨床情報、サンプル情報を結合するために必要になる。ここでは腫瘍組織由来のデータフレームsample_tumor_mergedと、正常組織由来のデータフレームsample_normalで、今後、全データフレームを結合するための軸になりそうな列である列cases.case_idと列samples.sample_idについて、データフレームsample_tumor_mergedとデータフレームsample_normalで互いに重複した行は無いことを%in%を使って確認する。
ここでも、以前の解析ではなかった、列cases.case_idや列samples.sample_idというように、列の頭にドットで区切られた謎の接頭辞(cases.やsamples.)が付いている。こっちの方がわかり易いのだろうか…こうやって分類するにしたって、その分類が他のデータとどのように関連している(ひも付いている)かイマイチわからないので、無駄だとおもうのだが….というか、変数なんだからアンダーバーで良くないか?付けるならば「接頭辞を付けました。これはこのような基準で付けております。これらを元にして、各データセットを関連付けることができます。」くらいのシステムにしろよって感じである。そうじゃないなら、急に余計なことはしないでほしい。
# code 18
ncol(sample_tumor_merged) # 39
nrow(sample_tumor_merged )# 38229
ncol(sample_normal) # 39
nrow(sample_normal) # 5680
sample_tumor_merged$cases.case_id %in% sample_normal$cases.case_id %>% table() # All false
sample_tumor_merged$samples.sample_id %in% sample_normal$samples.sample_id %>% table() # All falseデータフレームsample_tumor_mergedとデータフレームsample_normalで、被っている症例はなさそうだったので、この2つのデータフレームを結合させてデータフレームsample_mergeを作成する。
# code 19
sample_merged <- rbind(sample_tumor_merged, sample_normal)
ncol(sample_merged) # 39
nrow(sample_merged )# 43909 = 38229 +5680作成したデータフレームsample_mergedの整理を行う。ここでも謎の欠損値「’–」をRの欠損値であるNAで置き換える。
謎の接頭辞はいいとして、その後ろのドットがどうしても邪魔なので、それをアンダーバーに置き換えようと思う。まず、データフレームsample_mergedからその列名を抜き出してきてデータフレームとし、その列名を列columnとして、これらをデータフレームcolumn_name_sample_mergedとする。そして、gsub()でその列columnに入っている文字列(これがデータフレームsample_mergedの列名であるが)の中のドットをアンダーバーに変換し、それをデータフレームcolumn_name_sample_merged_2とする。その値をデータフレームsample_mergedの列名として入れる。
ここで一応、どんなサンプルがあるのか、正常組織と腫瘍組織はどのくらいあるのかなど、table()で確認しておく。table()による列samples_sample_typeの集計結果をみていると、Metastatic、Additional Metastatic、Recurrent Tumor等もいくらか含まれていることがわかる。もちろん、これらの方が圧倒的に命に関わってくるし、創薬の一番最初のターゲットはMetastaticやRecurrent Tumor等になるので、これらとの比較などは非常に有用と思う。ただし、対象疾患を選ぶためには、どうしてもPrimary Tumorを解析せざるを得ないのも事実であり、結果を単純なものにするためにもこれらは除かなくてはならない。なので、今後のためにもデータフレームsample_mergedを列samples_tissue_typeで腫瘍(データフレームsample_merged_2_tumor)と正常(データフレームsample_merged_2_normal)に分ける。
# code 20
# Clean up sample
# Replace "'--" to "NA".
sample_merged[sample_merged == "'--"] <- NA
# Check number of case
nrow(sample_merged) # 38229
# If peripheral blood, bone marrow, and tissue were collected from a patient, the dataset "sample_merge" contains 3 records for each individual.
# Replace "." into "_" in column names of dataset "sample_merged".
column_name_sample_merged <- colnames(sample_merged) %>% data.frame() %>% rename("column" = ".")
column_name_sample_merged_2 <- gsub("\\.", "_", column_name_sample_merged$column)
view(column_name_sample_merged_2)
colnames(sample_merged) <- column_name_sample_merged_2
column_name_sample_merged_2 <- data.frame(column_name_sample_merged_2) %>% rename("column" = "column_name_sample_merged_2")
# Check what kind of speciemens are in dataset "sample_merged".
table(sample_merged$samples_sample_type) %>% data.frame() %>% view()
# Check what kind of specimens are in dataset "sample_merged". At first, looking at "tissue_type" suggest that there are information of tumor and normal tissue (probably) surrounding tumor.
table(sample_merged$samples_tissue_type)
# 2025 10 24
# There are 14806 normal tissues, and 23423 tumor tissues.
# In this analysis, Tumor and Normal tissue are interested in.
sample_merged_2_tumor <- sample_merged %>% filter(samples_tissue_type == "Tumor")
sample_merged_2_normal <- sample_merged %>% filter(samples_tissue_type == "Normal")データフレームsample_merged_2_tumorの列samples_sample_typeがPrimary Blood Derived Cancer – Peripheral BloodもしくはPrimary Tumorのサンプルをfilter()して、データフレームsample_merged_3_tumorを作る。
このデータフレームsample_merged_3_tumorの列samples_submitter_idに関して、重複があるかどうかを確認する。列samples_submitter_idは、おそらくそのデータの提出元の機関と思う。そして各症例に関して列cases_case_idを見てみると、それらも同じである。違う値としては、例えば列samples_days_to_collection、列samples_pathology_report_uuid、列samples_preservation_method等が違っている。つまり、これらは同一症例から別の目的で採取されたサンプルであるらしい。色々考えたが、同一の症例である以上、RNA-seqのカウント値とこれらの症例を紐付けることができれば問題ないことがわかる。その症例の臨床情報とRNA-seqのカウント値が結びついていれば良い。よって、データフレームsample_merged_2_normalの列samples_submitter_idをdistinct()により、単に重複を除去し、データフレームsample_merged_4_tumorを作成する。本当ならば、列samples_days_to_sample_procurementや列samples_initial_weight等の値が有る症例を選んだほうが良いのであるが、これらは規則正しく入力されているわけではなく、これらを鑑みると非常にややこしくなるので、ここではそれはやらずに、無差別にdistinct()することにする。
# code 21
## The output suggested that several metastatic, FFPE, recurrent, and new primary tumor were included in the dataset. Our interest is "primary tumor" and therefore "Primary Blood Derived Cancer - Peripheral Blood" and "Primary Tumor" will be left.
table(sample_merged_2_tumor$samples_sample_type)
sample_merged_3_tumor <- sample_merged_2_tumor %>% filter(samples_sample_type == "Primary Blood Derived Cancer - Peripheral Blood" | samples_sample_type == "Primary Tumor")
# Check if column "sample_submitter_id", which holds the value like "TCGA-NC-A5HJ-01Z", in dataset "sample_merged_3" contains all unique or duplicated values.
checK_sample_submitter_id_tumor <- sample_merged_3_tumor %>% group_by(samples_submitter_id) %>% filter(n() > 1)
# There are 310 duplicated cases.
# In this analysis, purpose of dataset "sample_merge" is for the matching each case and count data. Therefore, I think de-duplication is good enough for further analysis. If you need, for example, pathological report (which is stored at column "pathlogy_report_uuid") or, you need to analyze the difference between sample collection days (which is stored at column "days_to_collection"), you need to get back here to check which cases are required for the analysis.
# Therefore, duplicated cases will be simply removed from the dataset "sample_merged_3".
# I think we need to use the information that are associated with RNA-seq count, which is linked with "file_name".
sample_merged_4_tumor <- sample_merged_3_tumor %>% distinct(samples_submitter_id, .keep_all = TRUE)次に、code 21と同じような処理をデータフレームsample_merged_2_normalに対しても行い、データフレームsample_merged_3_normalを作成する。
# code 22
# Cleanup sample_merged_2_normal
## The output suggested that several metastatic, FFPE, recurrent, and new primary tumor were included in the dataset. Our interest is "primary tumor" and therefore "Primary Blood Derived Cancer - Peripheral Blood" and "Primary Tumor" will be left.
table(sample_merged_2_normal$samples_sample_type)
# sample_merged_3_normal <- sample_merged_2_normal %>% filter(samples_sample_type == "Primary Blood Derived Cancer - Peripheral Blood" | samples_sample_type == "Primary Tumor")
# Check if column "sample_submitter_id", which holds the value like "TCGA-NC-A5HJ-01Z", in dataset "sample_merged_3" contains all unique or duplicated values.
checK_sample_submitter_id_normal <- sample_merged_2_normal %>% group_by(samples_submitter_id) %>% filter(n() > 1)
# There are 310 duplicated cases.
# In this analysis, purpose of dataset "sample_merge" is for the matching each case and count data. Therefore, I think de-duplication is good enough for further analysis. If you need, for example, pathological report (which is stored at column "pathlogy_report_uuid") or, you need to analyze the difference between sample collection days (which is stored at column "days_to_collection"), you need to get back here to check which cases are required for the analysis.
# Therefore, duplicated cases will be simply removed from the dataset "sample_merged_3".
# I think we need to use the information that are associated with RNA-seq count, which is linked with "file_name".
sample_merged_3_normal <- sample_merged_2_normal %>% distinct(samples_submitter_id, .keep_all = TRUE)
ここで、今後の解析で必要となるだろうデータフレームについてその症例を確認する。必要となるだろうデータフレームは、original_count、samplesheet_merged、clinical_merged_required_survival_de_duplicated、sample_merged_4_tumor、sample_merged_3_normalである。行は症例なので、nrow()で症例数を確認しておく。
# code 23
# Required dataset for further analysis.
# count dataset
original_count # this is original count data, which compose fine_name as column.
ncol(original_count) #14223 (14221 cases + gene_id + gene_name)
# samplesheet_merged
samplesheet_merged
nrow(samplesheet_merged) # 14221 cases
# clinical_merged_required_survival_de_duplicated
clinical_merged_required_survival_de_duplicated
nrow(clinical_merged_required_survival_de_duplicated) # 11696 cases
# sample_merged_4_tumor
sample_merged_4_tumor
nrow(sample_merged_4_tumor) # 22509 records, because there are several different information for a case.
# sample_merged_3_normal
sample_merged_3_normal
nrow(sample_merged_3_normal) # 14779 records, because there are several different information for a case. まずはデータフレームsamplesheet_mergedに対してarrange()を使い、他のデータフレームと結合するための軸となりそうな列の値で並び替える。ちなみに、このarrange()による並び替えは、もしかしたら不要かもしれない。最終的にinner_join()で結合するので、別に並んでなくても良い。ただし、何かと見にくいので並べ替えるのもあり、という感じである。ここでは、列project_idと列case_idを使用する。また、列samples_submitter_idの頭に付いている「samples_」は不要な謎分類なので、これをrename()により消し、データフレームsamplesheet_merged_2とする。
このデータフレームsamplesheet_merged_2には原発腫瘍以外も含んでいる可能性があるので、それを確認する。table()で列tumor_discriptionと列tissue_typeのクロス集計を出力し、それをデータフレームcheck_freq_samplesheetとして作成する。このとき、出来上がったデータフレームcheck_freq_samplesheetの列名がV1、V2、V3とかなっているはずなので、列名元のデータフレームと一致させるためにcolnames()を使って列tumor_discriptionと列tissue_typeをそして集計の結果出てくる列に列Freqとして名前を付ける。
このデータフレームcheck_freq_samplesheetの列tumor_discriptionには、Primary以外にもMetastaticやRecurrence等が有るらしい。上述した通り、この解析は原発腫瘍に着目したいので、これらは除いた方が良さそうである。なので、データフレームsamplesheet_merged_2から、原発腫瘍(列tumor_discriptionが「Primary」かつ列tissue_typeが「Tumor」のもの)と正常組織(列tumor_discriptionが「Not Applicable」かつ列tissue_typeが「Normal」のもの)のデータだけをfilter()を使って抽出し、新たにデータフレームsamplesheet_merged_3を作成する。
次にデータフレームsample_merged_4_tumorを処理する。ここでも、まずは各データフレームを結合するときの軸になりそうな列である、列project_project_id、列cases_submitter_id、列samples_submitter_idをarrange()により並び替えておく。色々と列を見ていると、列samples_submitter_idが各疾患毎に重複がなさそうである。そこで、列samples_submitter_idを使って、データフレームsamplesheet_merged_3とデータフレームsample_merged_4_tumorをinner_join()する。ここで、お互いのデータフレームに同じ列が含まれているので、どちらの列がどちらのデータフレームから来たのかをはっきりさせるために、inner_join()の値としてsuffix = c(“_sample”, “_samplesheet”)を入れておく。データフレームsample_merged_3_normalに対しても同様の処理をし、それぞれデータフレームsample_samplesheet_tumorとデータフレームsample_samplesheet_normalを作成し、それらを最後にrbind()で縦方向に結合し、データフレームsample_samplesheetを作成する。出来上がったデータフレームsample_samplesheetについても、今後重要になりそうな列である列project_id、列case_id、列samples_submitter_idの値でarrange()を使って並べ替えておき、症例数をnrow()で確認しておく。並べ替えと症例数の確認は、まぁ、気分である。こうしなければなんか安心できないためだ。以降も、結合する前のデータセットsamplesheet_merged_3とか、結合した後のデータセットsample_samplesheetの症例数が同じだけあるか、なども色々と確認しておく。mutate()を使って、新しく列checkを使ってまで、結合前と結合後の症例が一致しているのかを確かめた。
ここで、データフレームsample_merged_4_tumorとデータフレームsample_merged_3_normalの列samples_submitter_idに関しては頭の「samples_」は取らへんのかいって感じである。
# code 24
# At first, merge "samplesheet_merged" and "sample_merged_4" by column "sample_submitter_id".
# order the files
# each value looks like;
# samplesheet_merged$project_id; TCGA-ESCA
# samplesheet_merged$case_id; TCGA-IG-A97I
# samplesheet_merged$sample_submitter_id; TCGA-IG-A97I-01A
samplesheet_merged <- samplesheet_merged %>% arrange(project_id, case_id, sample_id)
samplesheet_merged_2 <- samplesheet_merged %>% rename("samples_submitter_id" = "sample_id")
# samplesheet_merged_2 contains the tumor type other than primary tumor. it should be removed because they are not interested in this analysis.
check_freq_samplesheet <- table(samplesheet_merged_2$tumor_discription, samplesheet_merged_2$tissue_type) %>% data.frame()
colnames(check_freq_samplesheet) <- c("tumor_discription", "tissue_type", "Freq")
# samplesheet_merged_3 contains tumor and normal cases.
samplesheet_merged_3 <- samplesheet_merged_2 %>% filter(
(tumor_discription == "Primary" & tissue_type == "Tumor") |
(tumor_discription == "Not Applicable" & tissue_type == "Normal")
)
# Match column "sample_submitter_id" in dtaset "sample_merged_4" and column "sample_submitter_id" in dataset "samplesheet_merged".
# sample_merged_4$project_id; TCGA-LUSC
# sample_merged_4$case_submitter_id; TCGA-NC-A5HJ
# sample_merged_4$sample_submitter_id; TCGA-NC-A5HJ-01Z
sample_merged_4_tumor <- sample_merged_4_tumor %>% arrange(project_project_id, cases_submitter_id, samples_submitter_id)
sample_merged_3_normal <- sample_merged_3_normal %>% arrange(project_project_id, cases_submitter_id, samples_submitter_id)
check_freq_sample_merged_4_tumor <- sample_merged_4_tumor %>% group_by(samples_submitter_id) %>% filter(n() > 1) # There is no duplication.
# Merged samplesheet_merged and sample_merged_4 by column "sample_submitter_id".
sample_samplesheet_tumor <- inner_join(sample_merged_4_tumor, samplesheet_merged_3, by = c("samples_submitter_id"), suffix = c("_sample", "_samplesheet"))
sample_samplesheet_normal <- inner_join(sample_merged_3_normal, samplesheet_merged_3, by = c("samples_submitter_id"), suffix = c("_sample", "_samplesheet"))
sample_samplesheet <- rbind(sample_samplesheet_tumor, sample_samplesheet_normal)
# then order the dataset.
colnames(sample_samplesheet) %>% data.frame() %>% rename("column" = ".") %>% view()
sample_samplesheet <- sample_samplesheet %>% arrange(project_id, case_id, samples_submitter_id)
nrow(sample_samplesheet) # 13478 cases
# check order
nrow(samplesheet_merged_3) # 13478 cases
nrow(sample_samplesheet) # 13478 cases
order_sample_submitter_id<- samplesheet_merged_3$samples_submitter_id
check_sample_submitter_id <- sample_samplesheet$samples_submitter_id
length(order_sample_submitter_id) # 13478 cases
length(check_sample_submitter_id) # 13478 cases
check <- samplesheet_merged_3$samples_submitter_id %in% sample_samplesheet$samples_submitter_id %>% data.frame() %>% rename("samples_submitter_id" = ".")
table(check$samples_submitter_id) # 13478 TRUE
# samplesheet_merged_3 contains tumor and normal cases.
check_sample_order <- data.frame(samplesheet = order_sample_submitter_id, sample_samplesheet = check_sample_submitter_id)
check_sample_order <- check_sample_order %>% mutate(
check =case_when(
samplesheet == sample_samplesheet ~ "OK",
TRUE ~ "NG"
)
)
table(check_sample_order$check) # 13478 cases are OK.
# check column "case_id_samplesheet"
check_unique_case <- sample_samplesheet %>% group_by(samples_submitter_id) %>% filter(n() > 1)
# there are different file name in a same case.
check_unique_file <- sample_samplesheet %>% group_by(file_name) %>% filter(n() > 1) # all unique files
サンプルの情報と臨床情報のデータフレームsample_samplesheetと、症例毎のカウント値のファイル名は一致している必要がある。そこで、データフレームsample_samplesheetから症例毎のカウント値のファイル名が入っている列file_nameを取ってきて、それをベクトルorder_file_nameとする。このベクトルを使って、症例毎のカウント値のデータフレームoriginal_countから、列gene_id、列gene_name、一致するファイル名の列を抜き出してくる。これにはselect()とall_of()を使うのが良い。これをデータフレームcount_reorderとする。
# code 25
# make sure order the dataset "original_count" based on the order of "file_name" in dataset "sample_samplesheet".
# this is for the ordering dataset "count" because the count matrix must be stored into anndata object and therefore order of file must be consistent with all dataset.
order_file_name <- sample_samplesheet$file_name
length(order_file_name) # 13478 cases
# reorder the dataset "original_count" based on the order of "file_name" in dataset "sample_samplesheet".
count_reorder <- original_count %>% select(c("gene_id", "gene_name", all_of(order_file_name)))
ncol(count_reorder) #13480 (13478 cases + gene_id + gene_name)
gc()
gc()続いてデータフレームsample_samplesheetと、臨床情報が保持されたデータフレームclinical_merged_required_survival_de_duplicatedを列cases_case_idを軸にしてinner_join()する。列の重複があるかもしれないので、どの列がどのデータフレームだったか判別できるようにinner_join()にsuffix = c(“”, “_clinical”)を入れておく。これでサンプルの情報と臨床情報を結合したデータフレームsample_samplesheet_clinicalが出来上がる。そのデータフレームでも重要そうな列でarrange()により並べ替えし、column_to_rownames()で列file_nameをこのデータフレームsample_samplesheet_clinicalの行名にする。これで、疾患毎のカウント値に紐づく臨床情報が出来上がりである。
# code 26
# merge sample_samplesheet and clinical_merged_required_survival_de_duplicated
sample_samplesheet_clinical <- inner_join(
sample_samplesheet,
clinical_merged_required_survival_de_duplicated,
by = c("cases_case_id" = "cases_case_id"),
suffix = c("", "_clinical")
)
sample_samplesheet_clinical <- sample_samplesheet_clinical %>% arrange(project_project_id, cases_case_id, samples_submitter_id)
sample_samplesheet_clinical <- sample_samplesheet_clinical %>% column_to_rownames("file_name")
sample_samplesheet_clinical <- sample_samplesheet_clinical %>% arrange(project_project_id, cases_case_id, samples_submitter_id)
症例毎のカウント値が収められたデータフレームcount_reorderを整えなければならない。まずは重複した遺伝子を除く。これまでと同じように、見たい列である列gene_nameをgroup_by()に入れ、それをfilter(n()>1)に渡して、1回より多く出現する遺伝子を見てみる。そうすると、どうやら110個の遺伝子名が重複しているらしい。その重複した遺伝子名は、データフレームdf_gene_nameに入れておいた。
このデータフレームdf_gene_nameを使って、症例毎のカウント値を入れたデータフレームcount_reorderから、%in%を使って重複した遺伝子のカウント値を取ってきて、それをデータフレームcount_reorder_QCとする。このデータフレームcount_reorder_QCを眺めていてわかるのが、カウント値が著しく低い遺伝子があること、ensembleの遺伝子ID(列gene_idの値)の末尾に「_PAR_Y」が付いている遺伝子、すなわち、Y染色体に由来する遺伝子があること、リボソーム関連の遺伝子(U3とかU8とか)が沢山あるらしい。ここでは先ず、Y染色体に由来する遺伝子を除き、新しくデータフレームcount_reorder_QC_1を作成する。これらが疾患に関連する可能性は大いに有る、というか、こちらの方が科学的には面白いかもしれないが、Y染色体以外に由来する遺伝子(正直、両者とも似たようなシークエンスだろうから、本当にどちら由来なのかはっきりわからない。もちろん、Y染色体に由来する遺伝子に特有の配列がマップされていて、それがカウントされているのであれば、Y染色体由来のリードなんだろうけど)もあるので、それらを解析に含めようと思う。また、この手のフィルタリングでよく使われる手段であるrowSums()、すなわち、列の合計を列sum_countとして、また、参考までにある遺伝子のカウント値が0の症例の全症例に渡る割合を列percent_zero_countとしてmutate()で追加する。そして、列sum_countが0ではない遺伝子を抽出し、データフレームcount_reorder_QC_2とする。これでもまだ重複があるはずなので、列gene_nameをgroup_by()にいれ、そして、重複する遺伝子のうち、最大のカウント値の方を選んできて、それをデータフレームcount_reorder_QC_3とする。そして、これまでと同じように列gene_nameをgroup_by()に入れ、それをfilter(n()>1)で1回より多く出現する遺伝子があるかを確認し、それが無いことを確認する。そして、その遺伝子を元のデータフレームcount_reorderから拾って来てデータフレームcount_reorder_QC_4とした。このデータフレームcount_reorder_QC_4が、元のデータフレームcount_reorderの中で重複している遺伝子のうち、Y染色体由来では無く、かつ、その重複の中でも最もカウント値の合計が高い遺伝子である。
この重複した遺伝子を除いた遺伝子から成るデータフレームを、それら以外の、そもそも重複していないデータフレームと結合する必要がある。まずは元のデータフレームcount_reorderから重複がある遺伝子を全除いたデータフレームcount_reorder_no_duplを作り、それにデータフレームcount_reorder_QC_4の遺伝子をrbind()で縦方向に結合して、データフレームcount_reorder_post_QCとした。以降、group_by()とfilter(n()>1)で列gene_nameの重複が無いことを確認し、列gene_idを除き、データフレームcount_reorder_post_QC_2とした。列gene_idは完全に固有の遺伝子IDなので、絶対に重複はない。遺伝子名が重複していても、この遺伝子IDが重複することはないので、QCに使った。カウントマトリックスを作成するときには不要である。というか、遺伝子IDでもGSEA出来るんだけど、多くのパッケージが遺伝子名を使っているし、遺伝子IDを使うといちいち変換しなければならなず、面倒くさい。ここで、後に使用するかもしれないので、このデータフレームcount_reorder_post_QC_2の列gene_nameをベクトルorder_gene_nameとして保持しておく。これは、後に遺伝子名の順番をデータフレーム間で一緒にするためである。特に、Anndataで保存するときには、このあたりの順番が揃っていなければエラーが出る(ように思うが、定かではない。エラーになった記憶がある。)
このデータフレームcount_reorder_post_QC_2を使って、実際に解析に使用するための症例毎のカウント値をマトリックス(ここではカウントマトリックスと呼ぶ)にする。ここでちょっとした注意がある。マウントマトリックスというだけあって、そのマトリックスの中に文字列が含む列は入れたくない。なので、データフレームcount_reorder_post_QC_2からselect(-one_of())で列gene_nameを除き、データフレームoriginal_count_matを作成する。オブジェクト名に「_mat」とか付いているけど、まだマトリックスではない。将来的にマトリックスにするので、そのように名付けている。そして、そのデータフレームoriginal_count_matのうち、サンプルと臨床情報を保持したデータフレームsample_samplesheet_clinicalの行名と一致する列、すなわち、サンプルと臨床情報があるRNA-seqのカウント値のファイル名を取ってくる。このときのデータフレームoriginal_count_matは、行名が遺伝子、列名が各症例のカウント値のファイル名になっているので、それを今後のGSEAで使用できるようにt()で転置させ、それをデータフレームoriginal_count_mat_tとする。これで、行が症例毎のカウント値のファイル名、列が遺伝子名になっている。
ここで、このデータフレームcount_reorder_post_QC_2を使って行の合計値である列rowSumsと、ある遺伝子のカウント値が0である症例の全症例に対する割合である列percent_zero計算しておく。
この例では、最初に、列gene_nameのみから成るデータフレームcount_reorder_post_QC_3を作成し、それに列rowSumsと列percent_zeroを計算して入れた。そしてcolumn_to_rownames(“gene_name”)でデータフレームcount_reorder_post_QC_3の行名を遺伝子名にした。これは後にAnndataオブジェクトのvarデータフレームとして使用する。ただし、のAnndataオブジェクトは完全にオプションで、かつ、この解析では使用しない。本当はこれを使って解析を進めたかったが、本来AnndataはPythonのパッケージであり、Rで使おうとすると非常に使い勝手が悪かったためである。
また、症例毎のカウント値に加えて、列rowSumsと列percent_zeroを含んだデータフレームも、トラブルシューティング用に作成しておきたかったので、データフレームcount_reorder_post_QC_4も追加で作成しておいた。実際の解析には使用しないけど。
# code 27
# check are duplicated gene names in dataset "count_reorder".
# There are a lot of duplicated genes (110 genes).
df_gene_name <- table(count_reorder$gene_name) %>% data.frame() %>% rename("gene_name" = "Var1") %>% filter(Freq > 1)
nrow(df_gene_name) # 110 duplicated genes
# check_gene_name <- count_reorder %>% group_by(gene_name) %>% filter(n() > 1)
# select the duplicated genes
count_reorder_QC <- count_reorder[count_reorder$gene_name %in% df_gene_name$gene_name == TRUE,]
# remove genes at chromosome Y
# "PAR_Y" means the PseudoAutosomal Regions (PAR) of chromosome Y.
# https://www.gencodegenes.org/pages/faq.html
count_reorder_QC_1 <- count_reorder_QC %>% filter(str_detect(gene_id, "_PAR_Y") == FALSE)
# calculate sum of count and percent of 0 over the cases for every genes.
count_reorder_QC_1 <- count_reorder_QC_1 %>% mutate(
sum_count = rowSums(count_reorder_QC_1[,3:ncol(count_reorder_QC_1)]),
percent_zero_count = apply(count_reorder_QC_1[,3:ncol(count_reorder_QC_1)], 1, function(x) (sum(x == 0)/length(x)))
)
# # sum(x == 0) returns the number of 0 in a row.
# # following is practice to count number of 0 in a row.
# test_df <- data.frame(a = c(0,0,0,1,1), b = c(0,0,1,1,1), c = c(1,0,0,0,1))
# sum(test_df[1,] == 0)
# sum(test_df[4,] == 0)
# Of the genes in dataset "count_reorder_QC_1", remove no gene count in any cases.
count_reorder_QC_2 <- count_reorder_QC_1 %>% filter(sum_count != 0)
# Of the genes in dataset "count_reorder_QC_2", leave genes with maximum rowSum value in each gene.
count_reorder_QC_3 <- count_reorder_QC_2 %>%
group_by(gene_name) %>%
filter(sum_count == max(sum_count))
# check if the genes in dataset "count_reorder_QC_3" are duplicated or not.
check_duplicated_genes <- count_reorder_QC_3 %>% group_by(gene_name) %>% filter(n() > 1)
nrow(check_duplicated_genes) # 0
# count_reorder_QC_1, _2, and _3 contain "sum_count" and "percent_zero_count" columns. These columns are not required for the analysis.
count_reorder_QC_4 <- count_reorder[count_reorder$gene_id %in% count_reorder_QC_3$gene_id == TRUE,]
# remove duplicated genes from dataset "count_reorder".
count_reorder_no_dupl <- count_reorder[count_reorder$gene_name %in% count_reorder_QC$gene_name == FALSE,]
check_duplicated_genes_most_part <- count_reorder_no_dupl %>% group_by(gene_name) %>% filter(n() > 1)
nrow(check_duplicated_genes_most_part) # 0
# merged all unique genes.
count_reorder_post_QC <- rbind(count_reorder_no_dupl, count_reorder_QC_4)
check_duplicated_genes_post_QC <- count_reorder_post_QC %>% group_by(gene_name) %>% filter(n() > 1) # There is no duplication.
nrow(check_duplicated_genes_post_QC) # 0
# remove gene_id
count_reorder_post_QC_2 <- count_reorder_post_QC %>% select(-one_of("gene_id"))
# store order of gene_name
order_gene_name <- count_reorder_post_QC_2$gene_name
# This is final count matrix, 2025 10 25
# original_count_mat <- count_reorder_post_QC_2 %>% column_to_rownames("gene_name") # I do not kwow why but it does not work.
rownames(count_reorder_post_QC_2) <- count_reorder_post_QC_2$gene_name # instead of cocolumn_to_rownames(), use this code
original_count_mat <- count_reorder_post_QC_2 %>% select(-one_of("gene_name"))
original_count_mat <- original_count_mat %>% select(all_of(rownames(sample_samplesheet_clinical)))
original_count_mat_t <- t(original_count_mat)
view(head(original_count_mat_t))
# check which genes are significantly low counts over all cases.
## This is used for anndata.
count_reorder_post_QC_3 <- data.frame(gene_name = count_reorder_post_QC_2$gene_name)
count_reorder_post_QC_3$rowSums <- rowSums(count_reorder_post_QC_2[,2:ncol(count_reorder_post_QC_2)])
count_reorder_post_QC_3$percent_zero <- apply(count_reorder_post_QC_2[,2:ncol(count_reorder_post_QC_2)], 1, function(x) (sum(x == 0)/length(x)))
count_reorder_post_QC_3 <- count_reorder_post_QC_3 %>% column_to_rownames("gene_name")
## This is used for dataframe.
## If some calculation for each genes, use this dataframe.
# count_reorder_post_QC_4 <- count_reorder_post_QC_2 %>% column_to_rownames("gene_name") # Again, I do not kwow why but it does not work.
count_reorder_post_QC_4 <- count_reorder_post_QC_2 %>% select(-one_of("gene_name"))
count_reorder_post_QC_4$rowSums <- rowSums(count_reorder_post_QC_4[,1:ncol(count_reorder_post_QC_4)])
count_reorder_post_QC_4$percent_zero <- apply(count_reorder_post_QC_4[,1:ncol(count_reorder_post_QC_4)], 1, function(x) (sum(x == 0)/length(x)))
これで、実際に解析に使用するデータフレームたちが作成出来た。
実際に使用するのは、データフレームoriginal_count_mat_t、データフレームsample_samplesheet_clinicalの2つである。非常に紛らわしくて申し訳ないことだが、データフレームoriginal_count_mat_t、データフレームsample_samplesheet_clinical、データフレームcount_reorder_post_QC_3は、Anndataオブジェクトの作成に使用するが、これはAnndataオブジェクトを作るだけのもので、実際に作ったAnndataオブジェクトは解析に使用しない。最初の計画では使う予定だったんだけど、RでAnndataオブジェクトが使いにくかったため、断念した次第である。
しかも、以下のコードでちょっと微妙なことをやっていることに気がついた。この時点でデータフレームoriginal_count_mat_tは、マトリックスになっていない。それにも関わらず、データフレームoriginal_count_mat_tをまたdata.frame()に入れてデータフレームoriginal_count_mat_t_dfを作っている。これは二度手間である。でも、まぁ、目的のデータフレームが作られているということは問題なかったということなので、それをそのまま使っていく。
そういうことで、話を戻すと、ここ必要なデータフレームは、データフレームoriginal_count_mat_t_df(実際はoriginal_count_mat_tと同じはず)、データフレームsample_samplesheet_clinicalの2つである。この2つをinner_join()をファイル名でinner_join()したいので、まずはこれらのデータフレームに対してrownames_to_column(“file_name”)を使って、行名を列file_nameとして戻す。そして、この列file_nameを軸にして、データフレームoriginal_count_mat_t_df、データフレームsample_samplesheet_clinicalをinner_join()により結合し、データフレームorigianl_df_w_infoを作成し、改めてcolumn_to_rownames(“file_name”)で列file_nameを行名に直す。これで症例毎のカウント値、サンプルと臨床情報を結合して1つのデータフレームにすることが出来た。一応、症例数と遺伝子数をnrow()とncol()で確認しておく。
# code 28
view(head(original_count_mat_t)) # for matrix
view(head(sample_samplesheet_clinical)) # for obs
view(head(count_reorder_post_QC_3)) # for var
# Matrix
nrow(original_count_mat_t) # 10017 cases
ncol(original_count_mat_t) # 59422 genes
# Observations
nrow(sample_samplesheet_clinical) # 10017 cases
ncol(sample_samplesheet_clinical) # 66 conditions
# Variables
nrow(count_reorder_post_QC_3) # 59422 genes
ncol(count_reorder_post_QC_3) # 2 variables
# matrix and observations can be simply combined.
## at first, original_count_mat_t needs to be converted into data frame
view(head(original_count_mat_t))
original_count_mat_t_df <- data.frame(original_count_mat_t)
view(head(original_count_mat_t_df))
nrow(original_count_mat_t_df) # 10017 cases
ncol(original_count_mat_t_df) # 59422 genes
## then conbine original_count_mat_t_df and sample_samplesheet_clinical
original_count_mat_t_df <- original_count_mat_t_df %>% rownames_to_column("file_name")
sample_samplesheet_clinical_df <- sample_samplesheet_clinical %>% rownames_to_column("file_name")
origianl_df_w_info <- inner_join(
original_count_mat_t_df,
sample_samplesheet_clinical_df,
by = "file_name"
) %>% column_to_rownames("file_name")
nrow(origianl_df_w_info) # 11626 cases
ncol(origianl_df_w_info) # 59491 genes (59422 genes + 69 conditions)
今後はこのデータフレームorigianl_df_w_infoを使って様々な値を可視化するが、グラフの横軸やカテゴリーの名前にいちいち「TCGA」と付いているのは、センスが無い。だから、ここで、列project_project_idの値から頭の「TGCA-」を取ってしまい、疾患名だけの列diseaseを作成した。また、各症例に対して1からの通し番号を振りたかったので、row_number()を使って行の数をかぞれ、それをmutate()で新しく列individual_orderとした。そして、作成した列individual_orderと列diseaseを結合させて列disease_individual_orderを作った。
# code 29
origianl_df_w_info <- origianl_df_w_info %>% mutate(
disease = gsub("^TCGA-*", "", origianl_df_w_info$project_project_id),
order = c(1:nrow(origianl_df_w_info)))
origianl_df_w_info <- origianl_df_w_info %>%
group_by(disease) %>%
mutate(individual_order = row_number(disease))
origianl_df_w_info <- origianl_df_w_info %>% unite(col = "disease_individual_order", c("disease", "individual_order"), sep ="", na.rm = FALSE, remove = FALSE)
一応、データフレームorigianl_df_w_infoが今後の解析本番に使用していくデータフレームなのだが、この時点でRのキャッシュに70GBくらいのデータが保持されている。実際には使用しないデータフレームも沢山あるので、ここでTSVとして出力し、それを別のRのコードで再度読み込んで本番の解析を行っていく。
データフレームorigianl_df_w_infoは行名がファイル名になっているので、それをrownames_to_column(“file_name”)で改めて列file_nameに入れ直して、write_tsv()でTCVファイルを保存する。code 27で作成した、何かあったとき用のQC用データフレームcount_reorder_post_QC_4も同様の方法により行名である遺伝子名を列gene_nameに直して保存する。
# code 30
# these are for survival analysis
origianl_df_w_info_write <- origianl_df_w_info %>% rownames_to_column("file_name")
count_reorder_post_QC_4_write <- count_reorder_post_QC_4 %>% rownames_to_column("gene_name")
write_tsv(origianl_df_w_info_write, file = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/origianl_df_w_info.tsv")
write_tsv(count_reorder_post_QC_4_write, file = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/count_reorder_post_QC_4.tsv")Anndataオブジェクトを作成(Optional)
この章で作成するAnndataオブジェクトは以降の解析には使用しないので、飛ばしてしまっても良い。
本当ならば、この解析ではAnndataオブジェクトを使って解析を進めたかった。Anndataは一般的にはシングルセルRNAシークエンス解析用のPythonパッケージである。Rでも移植されているおり、この解析では症例数が10000以上も有るので、似たような解析が出来るだろうと、踏んでいた。ところが、RでAnndataを使おうとするとRでPythonを実行するためのパッケージであるReticulateを使う必要があり、そして、実際に使ってみるとなんとも効率の悪いことをしなければならないようだった。だったらはじめからPythonを使うわって話である。
しかしながら、PythonはRよりも圧倒的にメモリのハンドリングが良く、計算が早い。このような大きなデータセットを扱う場合、Pythonの方が圧倒的に早く同様の解析を出来ると思う。実際、この先に特定の遺伝子発現や遺伝子セットのエンリッチメントスコアを疾患毎にバイオリンプロットで比較したり、それらと生存率の関連性を解析したりする訳だが、そのときは非常に大きなデータセットを扱う必要があるわけで、そこでRではかなり遅くなってしまう。統計はRの方が強く、有用なパッケージも多いから、重要な解析、例えばescapeやgvsaなんかはRを使い、その途中のデータの整形、中央値の検定、カプランマイヤー法による生存率解析とそれに続くログランク検定なんかはPythonを使う方が良いのかもしれない。scRNA-seqでは実際にscanpyで基本的な解析を行い、DEG(Differential Expressed Gene)の統計解析などではRを使うということをやるが、これと一緒である。こういう意味で、将来のためにAnndataオブジェクトを出力しておこうと思う。
Anndataオブジェクトには、公式のページが示すとおり、カウントマトリックスXを中心に据え、その行に患者(scRAN-seqでは細胞)、列には遺伝子発現が入る。そして患者の情報をobsデータフレーム、遺伝子に関わる値をvarデータフレームとして入れていくこれらのデータフレームは他にも任意のものを増やすことが出来、さらに、データの構造に依存しないようなデータも入れることが出来る。何が良いかと言えば、これまでの準備でも散々気にしてきたような、データの順番や並びを気にする必要がないことであり、また各値をデータフレームとして使える、つまり、pandasパッケージで取り扱い、値をseabornパッケージで可視化でき、データフレームを取り扱う他のパッケージでもそれらを解析出来るのが利点と言える。逆に言えば、最初にAnndataオブジェクトを作るときはデータの順番が全部一致していないといけない。
そういうことで、まずはデータフレームorder_obs、データフレームorder_varに、それぞれ行名(症例毎のカウント値のファイル名、つまり症例名)と列名(遺伝子名)をデフォルトの値として保持しておく。また、Anndataオブジェクトのカウントマトリックスの部分であるXに入る予定のデータフレームoriginal_count_matの行名及び列名を、それぞれデータフレームobs_name_of_Xとデータフレームobs_name_of_Xとして保持しておく。後々Anndataオブジェクトとしてこれらを使うときには、その並びが一致していなければならない(はず)ので、データフレームvar_name_of_Xとデータフレームorder_var、もしくは、データフレームobs_name_of_Xとデータフレームorder_obsをcbind()で横方向に結合させ、データフレームcheck_obsとデータフレームcheck_varを作成し、出来た列同士を比較して、それらが同じかどうか確認しておく。
# code 31
# dataset for obs, which means observation.
# "sample_samplesheet_clinical" will be "obs".
# rownames of "sample_samplesheet_clinical" is "file_name"
order_obs <- rownames(sample_samplesheet_clinical) %>% data.frame() %>% rename("file_name" = ".")
# dataset for var, which means variable.
# "count_reorder_post_QC_3" will be "var"
# rownames of "count_reorder_post_QC_3" is "gene_name"
count_reorder_post_QC_3
order_var <- rownames(count_reorder_post_QC_3) %>% data.frame() %>% rename("gene_name" = ".")
# check order of variables in "original_count_mat"
# "original_count_mat" will be X.
var_name_of_X <- rownames(original_count_mat) %>% data.frame() %>% rename("gene_name" = ".")
obs_name_of_X <- colnames(original_count_mat) %>% data.frame() %>% rename("file_name" = ".")
#
check_var <- cbind(var_name_of_X, order_var)
colnames(check_var) <- c("var_name_of_X", "order_var")
check_var <- check_var %>% mutate(
check = case_when(var_name_of_X == order_var ~ "OK",
TRUE ~ "NG")
)
table(check_var$check) # 59422 cases are OK.
#
check_obs <- cbind(obs_name_of_X, order_obs)
colnames(check_obs) <- c("obs_name_of_X", "order_obs")
check_obs <- check_obs %>% mutate(
check = case_when(obs_name_of_X == order_obs ~ "OK",
TRUE ~ "NG")
)
table(check_obs$check) # 10017 cases are OK.
# order of obs (observations) and var (variables) looks good. Anndata object will be prepared at next step.
そして、pythonでは欠損値はnanなので、is.na()でNAがTrueなのかどうか確認し、それがTrueところに「nan」を入れる。こうしなければ、次にAnndataオブジェクトを作成するときにエラーが出てしまう。
# code 32
# Anndata is basically python library, and they can not handle "NA". Therefore, "NA" must be replaced with "nan".
sample_samplesheet_clinical[is.na(sample_samplesheet_clinical)] <- "nan"
そして、AnnData()を使ってAnndataオブジェクトを作成する。Xに疾患毎のカウント値のデータフレームoriginal_count_mat_t、obsにサンプルや診療情報が保持されたデータフレームsample_samplesheet_clinical、varに各遺伝子のカウント値から計算したrowSumsの値やpercent_zeroが入ったデータフレームcount_reorder_post_QC_3を入れて、Anndataオブジェクトoriginal_anndataを作成する。これを、H5ADフォーマットで保存しておく。こうすれば、もしPythonで解析したほうがいいなぁってなった場合に、解析に使用するデータセットをpythonのanndataパッケージで開くことが出来る。
# code 33
# sample_samplesheet <- sample_samplesheet %>% column_to_rownames("file_name")
# "original_count_mat_t" was prepared at line ~598.
library(anndata)
original_anndata <- anndata::AnnData(
X = original_count_mat_t,
obs = sample_samplesheet_clinical,
var = count_reorder_post_QC_3
)
write_h5ad(original_anndata, "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/original_anndata.h5ad")最後にsessionInfo()は忘れない方が良い。いざというときにこの情報から環境を再現することも出来るかもしれないし、そもそも将来的に報告書に書く必要が出てくると思う。
# code 34
sessionInfo()
# done at 2025 10 28ここからのRのダウンロード(文章を読みたくない人用)2
取り扱うデータフレームのサイズが一々大きく、これまでの解析から引き続き解析しているとメモリを超絶に圧迫するので、それを少しでも軽減するためにRのファイルを変える。code 30でTSVファイルとして出力したorigianl_df_w_info.tsvをfread()で読み込んできて、データフレームoriginalを作成する。そしてmutate()を使って、全部の行に「case」という文字列が入った列caseを作成し、次にunite()を使って列caseと列file_nameを結合し、新しく列caseを作成する。この列file_nameには、症例毎の通し番号が入っているので、この処理でcase1からcase12931までの値が入った列caseが出来上がる。
# code 34
# original <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/output/origianl_df_w_info.tsv")
# variable <- read_tsv("/mnt/seqdata/public_data/Blog/TCGA/output/count_reorder_post_QC_4.tsv")
# primary
original <- fread(
input = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/output/origianl_df_w_info.tsv",
sep = "\t",
encoding = "UTF-8",
data.table = FALSE) # This data must be used for the analysis
original <- original %>% mutate(case = "case")
original <- original %>% unite(col = "case", c("case", "file_name"), sep = "", remove = TRUE)
# IMPORTANT NOTICE column "case" will be used for a main key for inner_join() after several enrichment analysis (ssGSEA2, escape, gsva, fgsea)以下で、TCGAやCPTACでの各プロジェクトが一体何例くらいあるのかを確認する。
# code 35
table(original$project_project_id)
table(original$project_id)
table(original$project_project_id_clinical)CPTACは将来の解析で必要になるが、この解析では不要である。TCGAとCPTACの症例がどのくらいあるかを確認し、解析対象からCPTACを除外したデータフレームoriginal_tcgaを作成した。
# code 36
# Check case number for each project
original %>% filter(str_detect(project_project_id, "TCGA") == TRUE) %>% nrow() # 10967
original %>% filter(str_detect(project_project_id, "CPTAC") == TRUE) %>% nrow() # 1964
# prepare tcga
original_tcga <- original %>% filter(str_detect(project_project_id, "TCGA") == TRUE)
table(original_tcga$samples_tissue_type)escapeとGSVAを用いたエンリッチメント解析
GMTファイルのダウンロードと読み込み
ここで、GSEAの準備を始める。まず、必要なデータが、GMTファイルである。GMTファイルに関してはここに詳しく書いてある。簡単に言えば、何の分子経路(ここでは「遺伝子セット」と呼ぶ)にどんな遺伝子が含まれているか、を記載したテキストファイルである。今回はBroad Instituteが管理する遺伝子セットをまとめたデータベースであるMSigDBに登録のあるヒトの遺伝子セットを全部使うことにする。
ここで要注意点がある。ここに、以下のように書いてある。
NOTE: we strongly discourage running analyses against the full Human MSigDB GMTs. We recommend using the above GMTs instead for more focused results.
https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp
「全部使うのではなく、上記の遺伝セットを使ってより絞った結果を出せ」ということが書いてある。確かに、最初の注意点1に書いたように、全遺伝子セットを使うとかなりの計算時間になり、場合によっては計算が終わるまで数ヶ月も費やす、とかも、考えられる。そういう場合は当然ながら考える必要が有る。でも、別にやるなとは書いてない。だから、コンピューターリソースが許す限りやったらいいと思う。ここからヒトの遺伝子セットを全部ダウンロードしてきて、それを使用する。バージョンはv2025.1である。

ダウンロードしたら、それを適宜展開し、GSVAパッケージのreadGMT()を使ってGMTファイルを読み込んで、それをオブジェクトmsigdb_2025_1にする。
# code 37
msigdb_2025_1 <- readGMT("/mnt/team4tb/Dropbox/Blog/2024 11 02 TCGA analysis/msigdb_v2025.1.Hs_files_to_download_locally/msigdb_v2025.1.Hs_GMTs/msigdb.v2025.1.Hs.symbols.gmt")
escapeとGSVAで使用するマトリックスの作成
データフレームoriginal_tcgaから、症例毎のカウント値に相当するところ(1列目から59423列目)を取ってきて、症例毎の通し番号の列caseの列名をrename()でNAMEに変え、それをcolumn_to_rownames()で行名にする。こうしないと、t()で転置したときに行名が消えてしまうためである。この時点で行が症例、列が遺伝子になっているので、それをt()で転置させて行が遺伝子、列が症例にして、データフレームenrich_3_tとする。escapeやGSVAには、このデータフレームenrich_3_t(実際にはそれをマトリックスに直したマトリックスenrich_3_t_mat)を使用する。そして、rownames_to_column()により改めて行名を列NAMEとして戻し、それをデータフレームenrich_3_t_1とする。このデータフレームenrich_3_t_1は、この後に作成するGCTファイルに必要である。
実際のescapeとGSVAでは値としてマトリックスを使う必要がある。データフレームenrich_3_tをas.matrix()でマトリックスenrich_3_t_matに変換する。ついでに、データフレームenrich_3もas.matrix()に入れて、マトリックスenrich_3_matを作っておいた。
# code 38
enrich_1 <- original_tcga[, 1:59423] #
enrich_2 <- enrich_1 %>% rename("NAME" = "case")
enrich_3 <- enrich_2 %>% column_to_rownames("NAME")
enrich_3_t <- t(enrich_3) %>% data.frame()
enrich_3_t_1 <- enrich_3_t %>% rownames_to_column(var = "NAME") # this is for preparation of GCT file.
#
enrich_3_t_mat <-as.matrix(enrich_3_t) # this is for various enrichment analysis.
enrich_3_mat <- t(enrich_3) # this is for various enrichment analysis.
view(enrich_3_t_mat) # just checking the data.
view(enrich_3_mat) # just checking the data.
gc()
gc()
#GCTファイルの作成(Optional)
今回はescapeとGSVAを使用するので、実際にはGCTファイルは使わないのだが、その途中に作成するマトリックスは必要である。もし遺伝子セットの数が少ない場合などはssGESA2も使用出来る。こういう場合はGCTファイルが必要になる。せっかくだから、GCTファイルの作り方も述べておく。GCTファイルについてはここに詳しく書いてある。GCTファイルの1行目は「#1.2」を入れる。2行目に遺伝子数(行数)、タブ、検体数(列数)を入れる。3行目の1列目が遺伝子名、3行目の2列目から検体(症例)毎のカウント値を入れていく。これはssGSEA2を使う場合のGCTファイルである。Broad InstituteのGSEAを行う場合のGCTファイルは、2行目までは一緒で、3行目の1列目が遺伝子名、3行目の2列目が「Description」、3行目の3列目から検体(症例)毎のカウント値になる。この場合の「Description」は何を入れても良いが、おすすめは遺伝子名に対応するEnsenblの遺伝子IDが良い。
GCTファイルはこのようにちょっと意味わからんフォーマットなので、cat()で一行ずつ出力していくのが良いと思っている。いつも自分はcat()とpaset0()とwrite_tsv()を使ってGCTファイルを作っている。注意点としては、cat()でもwrite_tsv()でも、元から有る同じファイル名のファイルに、引数append = TRUEを付けることで、内容をどんどん付け足していく。なので、これをもう一度流すときは最初にfile.remove()で、元のファイルを一旦削除してから、改めてcat()していく。以下で作成したGCTファイルは、ssGSEA2用で使用することが出来る。だからといって今回の解析ではこれは使用しない。遺伝子数、症例数、遺伝子セット数のどれもかなり多く、計算時間が膨大になるはずだし、正直、個人的にはGSVAの結果が一番使いやすいためである。
# code 39
header <- "#1.2"
number_gene <- nrow(enrich_3_t_1) # 59422 genes
number_sample <- ncol(enrich_3_t_1)-1 # 10017 cases
file.remove("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/TCGA.gct")
cat(header, "\n", file = "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/TCGA.gct")
cat(number_gene, "\t", number_sample, "\n", file = "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/TCGA.gct", append = TRUE)
write_tsv(enrich_3_t_1, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/TCGA.gct",
col_names = TRUE,
append = TRUE)escapeによるエンリッチメント解析
code 38で作成したマトリックスenrich_3_t_matと、ダウンロードしたGMTファイルを使ってescapeパッケージによりエンリッチメント解析を行う。計算に費やした実際の時間がきになるので、取り掛かった時間と計算が終わった時間をSys.time()で測定しておいた。計算結果はオブジェクトresult_escapeに入れる。計算時間は、数分で終わる。
# code 40
# escape, msigdb
set.seed(1)
start_results_escape <- Sys.time()
result_escape <- escape.matrix(
input.data = enrich_3_t_mat,
gene.sets = msigdb_2025_1,
method = "ssGSEA",
groups = 1000,
min.size = 10)
end_results_escape <- Sys.time()
gc()
gc()GSVAによるエンリッチメント解析
escapeと同様に、マトリックスenrich_3_t_matと、ダウンロードしたGMTファイルを使ってGSVAパッケージによりエンリッチメント解析を行う。計算結果はオブジェクトresult_gsvaに入れる。この計算時間も数分で終わる。
# code 41
# geva, msigdb
set.seed(1)
start_results_GSVA <- Sys.time()
gsvaPar <- gsvaParam(enrich_3_t_mat, msigdb_2025_1)
result_gsva <- gsva(gsvaPar, verbose=TRUE)
end_results_GSVA <- Sys.time()
gc()
gc()エンリッチメント解析の結果をまとめる
最終的には、エンリッチメント解析の結果をデータフレームoriginal_tcgaに結合したいので、行を症例、列をエンリッチメントスコアにしたい。escapeの結果はこの形で出力されるので、そのままデータフレームに直せば良い。まずは、列名を付ける。列名を付けるために、まずは出力結果であるresult_escapeからその列名(遺伝子名)を取ってきて、それをデータフレームcolumn_escapeに直し、さらの列名をgenesetに直す。そのデータフレームcolumn_escapeに、文字列escapeという列library(packageの方が望ましい。用語的に。)をmutate()を使って追加する。そして、列genesetと列libraryを結合し、新しく列genesetと名付ける。この列genesetをescapeの結果であるマトリックスresult_escapeの列名とし、さらに、後のinner_join()のためにデータフレームresult_escapeに直し、rownames_to_column()を使ってその行名をcaseにしておく。
# code 42
# escape
column_escape <- colnames(result_escape) %>% data.frame()
colnames(column_escape) = "geneset"
column_escape <- column_escape %>% mutate(library = "escape")
column_escape <- column_escape %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_escape) <- column_escape$geneset
result_escape <- as.data.frame(result_escape) %>% rownames_to_column(var = "case")次に、GSVAの結果を整える。GSVAはescapeと異なり、行がエンリッチメントスコア、列が症例として出力される。なので、先ずはt()を使って転置させ、行が症例、列がエンリッチメントスコアにする。以降はescapeの場合と同様の処理でデータフレームresult_gsvaを作成する。
# code 43
# gsva
## prepare dataset
result_gsva_t <- result_gsva %>% t() %>% data.frame()
## prepare column name, hallmark
column_gsva <- colnames(result_gsva_t) %>% data.frame()
colnames(column_gsva) = "geneset"
column_gsva <- column_gsva %>% mutate(library = "gsva")
column_gsva <- column_gsva %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_gsva_t) <- column_gsva$geneset
result_gsva <- result_gsva_t %>% rownames_to_column(var = "case")
escapeとGSVAの結果をリストに入れ、reduce()とinner_join()で一気に結合し、データフレームanalysisを作成する。このあたりから、データフレームのサイズが少なく見積もって2倍ずつ増えていき、その計算のためにさらに大きなメモリが使用されるので、単純な操作でもけっこうな長時間待たされるようになるところが注意点である。
# code 44
# merge all dataset
temp <- list()
temp[[1]] <- original
temp[[2]] <- result_escape
temp[[3]] <- result_gsva
analysis <- reduce(temp, inner_join) %>% data.frame()ヒストグラムとバイオリンプロットによる疾患毎の遺伝子量またはエンリッチメントスコアの比較
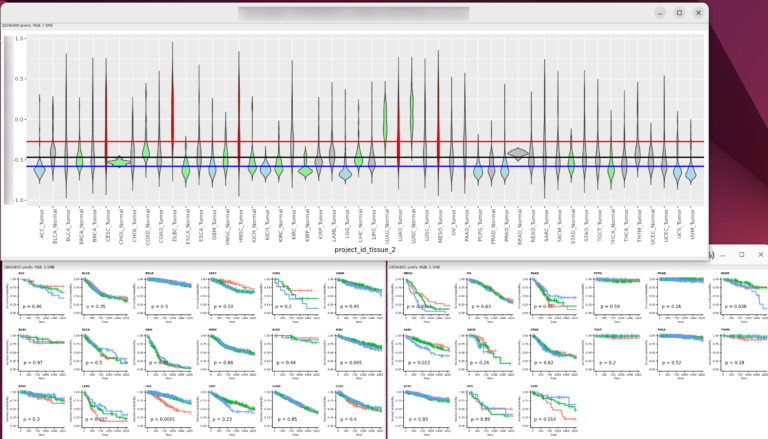
今回は正常組織と腫瘍組織の両方があるので、特にバイオリンプロットに両者の比較の統計値(Wilcoxinの順位和検定;Mann-WhitneyのU検定)を行った結果を反映しようと思う。まずは基本のヒストグラムから作成する。ここで示すヒストグラムは1次元的で、遺伝子発現やエンリッチメントスコアが全症例に渡ってどういった分布をしているのかくらいしかわからない。主に、何らかのQCのためにデータを確認する目的に使用できると思う。
データの準備
最初に、table()でデータの軸になりそうな値をざっと眺める。データフレームanalysisの列samples_tissue_typeには組織が正常か腫瘍か、列project_project_id_clinicalには、値として「TCGA-ACC」のように「TCGA-疾患」という値が入っている。ここでは、unite()を使ってこの2つの列を結合し、列project_id_tissueを作り、その列を含むデータフレームviolinを作る。
ここで、次の処理のために、table()で列project_id_tissueを集計し、その結果をデータフレームtable_project_id_tissueとして作成する。
# code 45
# Check values
table(analysis$samples_tissue_type)
table(analysis$project_project_id_clinical)
table(analysis$project_project_id_clinical, analysis$samples_tissue_type)
# Prepare dataset for graph
violin <- analysis %>% unite(
col = "project_id_tissue",
c("project_project_id_clinical", "samples_tissue_type"),
sep = "_", remove = FALSE)
table_project_id_tissue <- table(violin$project_id_tissue) %>% data.frame()
同様に、データフレームviolinで使えそうな列をtable()を使って集計する。
また、# code 45で作成したデータフレームtable_project_id_tissueを使って、あまりにも例数の少ない正常組織のカウントを除いてしまうことにした。ここでは例数が5より低い組織を除いた。この理由は、後のバイオリンプロットを使った疾患毎の遺伝子発現量もしくはエンリッチメントスコアの比較のとき、実際はそうでもないのに正常組織に比べて腫瘍組織が顕著に高いもしくは低い印象があったためである。バイオリンプロットは例数が3とかでもなんとなく分布を持ったような形で表示されるため、このような過大評価の印象を与えてしまうことに気がついた。そもそも、正常組織が3や、たとえ10だったとしても、それに対する腫瘍の数が150とかとはあまり釣り合っていないような気がする。統計値は出るのかもしれないけど、正しいとは思えない。だから、5より少ない例数の組織を除くことにした次第である。
そういうことなので、例数が5より低い正常組織はデータフレームtable_project_id_tissue_must_be_removedに入れてあるので、%in%を使ってそれ以外をデータフレームviolinに残す。
次に、データフレームviolinの列project_id_tissueから、文字列の頭に付いている「TCGA-」を除いて、新しく列project_id_tissue_2を作る。これで、例えば、BRCA_NormalとかBRCA_Tumorとかのように、ある疾患とそれに対する正常組織をパッと区別できるようになる。というか、別に「TGCA-」が頭に付いていてもいいのだが、やっぱり図とかの作法として分かりきった事は除くべきだし、そもそも、横軸全部の頭に「TCGA-」が付くのは不細工だと思う。
そして、この列project_id_tissue_2に、factor()で順序を入れる。これで、ヒストグラム用のデータは出来上がりである。
# code 46
# Check values
table(violin$samples_tissue_type)
table(violin$project_project_id_clinical)
table(violin$project_project_id_clinical, analysis$samples_tissue_type)
table(violin$project_id_tissue)
# Several normal tissue cases are a very few. These cases are lilely to be too less for statstics. Therefore, they will be removed.
table_project_id_tissue_must_be_removed <- table_project_id_tissue %>% filter(Freq < 5)
# prepare dataset after removing few normal tissue cases
violin <- violin %>% filter(!project_id_tissue %in% table_project_id_tissue_must_be_removed$Var1)
# Check values
table(violin$project_id_tissue)
# release unused memory
gc()
gc()
violin <- violin %>% mutate(
project_id_tissue_2 = gsub(pattern = "^TCGA-", replacement = "", x = violin$project_id_tissue))
# Add order for graph
violin$project_id_tissue_2 <- factor(
violin$project_id_tissue_2,
levels = names(table(violin$project_id_tissue_2)),
labels = names(table(violin$project_id_tissue_2)),
exclude = NA,
ordered = is.ordered(violin$project_id_tissue_2),
nmax = NA)
table(violin$project_id_tissue)
names(table(violin$project_id_tissue))
# release unused memory
gc()
gc()ヒストグラムを表示する
ggplot2のヒストグラムで、エンリッチメントスコアHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaを表示する。これがGSVAによる結果である。
# code 47
# histogram
# gvsa
ggplot(violin, aes(x = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
geom_histogram(binwidth = 10*(1/nrow(violin))) +
xlim(range(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva))
median(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva) # -0.4754158
range(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva) # -0.8050957 0.6012628
quantile(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75)) # 1.061641 1.088336 1.116298 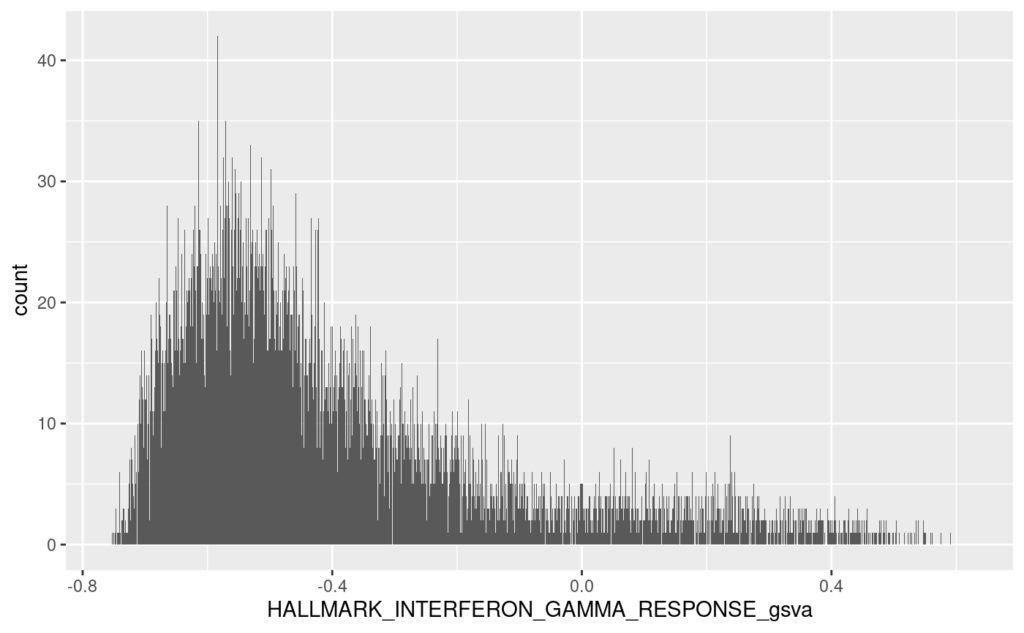
そして、これがescapeによるエンリッチメントスコアHALLMARK_INTERFERON_GAMMA_RESPONSE_escapeのヒストグラムである。
# code 48
# histogram
# escape
ggplot(violin, aes(x = HALLMARK_INTERFERON_GAMMA_RESPONSE_escape)) +
geom_histogram(binwidth = 10) +
xlim(range(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape))
median(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape) # -0.4754158
range(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape) # -0.8050957 0.6012628
quantile(violin$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape, probs = c(0.25, 0.5, 0.75)) # 1.061641 1.088336 1.116298 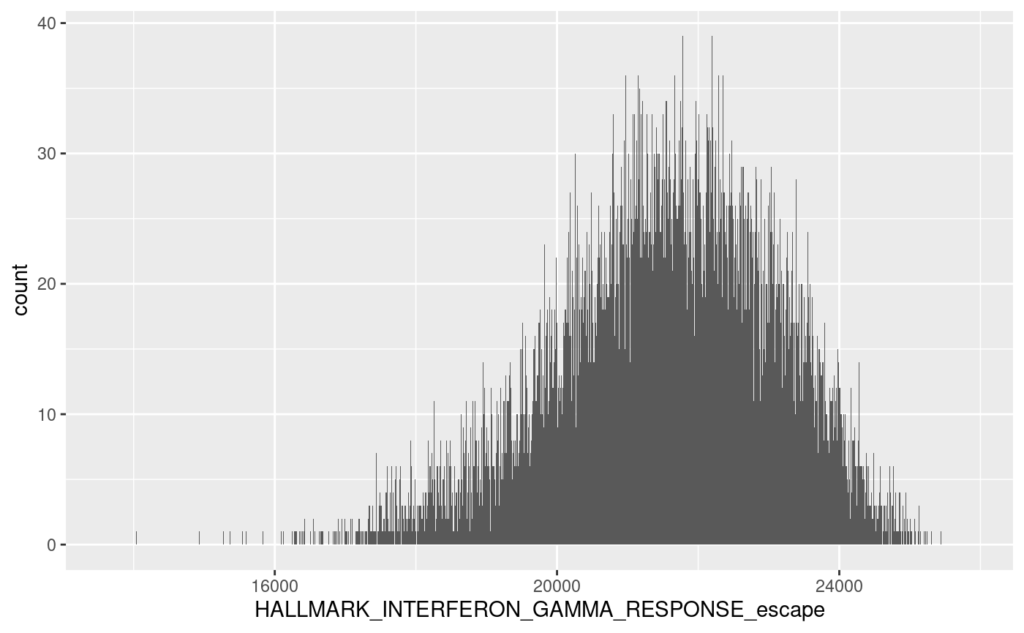
escapeの方がばらつきが少ないように見えるが、おそらくescapeの方も横軸を対数にすれば同じような分布になるのではないかと思っている。GSVAのほうが分布が広く階層化しやすそうだし、変に対数に変換しても後の計算がおかしくなっていたら嫌なので、GSVAを使っていく。ただし、値がマイナスっていうところはどのように影響してしまうのか、ちょっとわからんので注意が必要だと思う。
バイオリンプロットの準備
まずはじめに、以降の計算における注意点を述べる。ここから、リストを使って各遺伝子もしくは各遺伝子セットに対して計算を行っていくが、ここではシンプルなfor()を使っていくので、一つのコードチャンクにもけっこうな時間を要する場合がある。さらに、ここまでの処理と保持するデータセットがメモリをかなり圧迫しているので、それも計算をさらに遅くしているように思う。
疾患毎の遺伝子発現量、エンリッチメントスコアをバイオリンプロットで比較する。今回は、その登録が有る場合は正常組織の遺伝子発現量も加える。これにより、着目する遺伝子の発現量が、どの疾患で高いのか、それとも低いのか、正常組織と比べてどうかを検討出来る。バイオリンバイオリンプロットには、正常組織を除く疾患全体の25パーセンタイル、50パーセンタイル(中央値)、75パーセンタイルを示す水平線をそれぞれ青、黒、赤で入れる。また、コード内の一箇所に遺伝子もしくは遺伝子セットを入力すれれば済むようにする。
まずは、データセットを準備してパーセンタイルを計算する。最初にデータフレームviolinから疾患のデータだけを選んできて、データセットtumorを作成する。着目するのは「ある遺伝子の発現量が、どの疾患で高いか、低いか」なので、ここには正常組織は入らない。そして、このデータセットtumorの各遺伝子のカウント値、各遺伝子セットのエンリッチメントスコアを一つずつリストに入れていく。カウント値とエンリッチメントスコアだけでなく、分類するための列も取ってきてそのリストに入れておく。ここでは1列目の列case、59494列目の列disease_individual_order、59495列目の列disease、59460列目の列project_id_tissue_2、123824列目の列samples_tissue_type、そして、2列目から59423列目までの遺伝子名をリストgeneに入れ、そのリストgeneの各要素にname()で遺伝子名を付ける。遺伝子セットでも同様の処理をして、リストenrichmentを作成する。遺伝子セットは列59498目から列123823目にある。それらリストgeneとリストenrichmentをc()で縦方向に結合し、リストstatsを作成する。また、これと同様の処理をデータフレームviolin、すなわち、正常組織も含むデータでも行う。計算にはパーセンタイル等の計算には腫瘍だけのデータフレームtumorを使うが、バイオリンプロットでは疾患とそれに対する正常組織の遺伝子発現量もしくはエンリッチメントスコアを表示したいためである。上記にならって、リストgene_w_normal、リストenrichment_w_normal、そしてリストstats_w_normalを作成する。
次に、疾患全体の25パーセンタイル、50パーセンタイル、75パーセンタイルを計算する。そのためにまず、リストQ25_all、Q50_all、Q75_allを定義し、code 49で作成したリストstatの6列目、すなわち、各遺伝子のカウント値もしくは各遺伝子セットのエンリッチメントスコアを使って、疾患全体の 25パーセンタイル、50パーセンタイル、75パーセンタイルを計算していく。各リストにはnames()で遺伝子名もしくは遺伝子セット名を付ける。
続いて、各疾患の25パーセンタイル、50パーセンタイル、75パーセンタイルを計算する。これは、シンプルにgroup_by()で列diseaseをグループ化して、それで計算すればいいかと思っていたけど、どうやらそれだと上手く行かなかった。だから、ここでは、.dataという参照方法を使用する。本来ならば直接データフレームを参照するが、この.dataによりデータフレームを変数を介して参照出来る。ここに詳しく書いてある。rlangとdplyrで使えるようだ。まずは各遺伝子もしくは各遺伝子セットのデータをデータフレームdfに入れ、また、そのデータフレームdfの名前、すなわち遺伝子名もしくは遺伝子セット名をnames()で長さが1のベクトルgenesetに入れる。そして、そのデータフレームdfの列diseaseをgroup_by()でグループ化し、それぞれのグループ、すなわち、データフレームdfに入っている疾患毎に各統計量を計算していく。ここでも.data[[geneset]]という参照方法を使用する。長さが1のベクトルgenesetには、例えば「TP53」などのような遺伝子名を示す文字列が入っているわけだが、この参照方法により、TP53という名前のデータフレームを動的に参照することが出来るっていうわけらしい。こういったfor()でいちいち参照するデータフレーム名と列名が変わっていくような場合はどうやら有効のようだ。最初はstats[[i]][,6]とかを使えば良いと思ってたんだけど、これだとエラーで進まなかった。統計値をひとしきり計算したら、それをそのままmutate()に渡して、疾患毎のパーセンタイルと疾患全体のパーセンタイルを比較して階層化してしまう。各疾患の50パーセンタイルが疾患全体の25パーセンタイル以下ならば「0_low」、各疾患の50パーセンタイルが疾患全体の75パーセンタイル以上ならば「2_high」、各疾患の50パーセンタイルが疾患全体の25パーセンタイルより高くかつ75パーセンタイルより低い場合は「1_even」という階層にし、それを列colorにいれた。最後に出来上がったリストstats_2にリストstatsと同じ名前を付ける。この階層が、バイオリンプロットで各疾患を表示したときの色を示す値に使用と思う。つまり、列colorが2_highの疾患は赤、0_lowの疾患は青、1_evenの疾患は灰色というようにしようと思う。
ここ、リストstats_2に入れた列colorをリストstats_w_normalにも反映させる。match()を使って、リストstats_2の列diseaseとリストstats_w_normalの列diseaseが一致しているかを判別するデータフレーム(これにはTrueとかFalseとかのブール値が入っているはず)は入ったリストcolor_matchingを作成する。リストstats_w_normalの各要素の列diseaseが、リストstats_2の各要素の列diseaseと一致している場合、そのリストstats_2の各要素の列colorの値をリストstats_w_normalの各要素の列colorに入れていき、これをfor()で各要素に適用していく。
ここで、どの遺伝子もしくは遺伝子セットをバイオリンプロットで表示するか選ぶために、リストstats_w_normalの各要素の名前、すなわち、遺伝子名と遺伝子セット名だけを入れたデータフレームgene_geneset_nameを作成する。これで遺伝子名もしくは遺伝子セット名をチェックしようと思う。
# code 49
# violin plot
## prepare data
### tumor only
# Prepare dataset for quantile of tumor
tumor <- violin %>% filter(samples_tissue_type == "Tumor")
# release unused memory
gc()
gc()
#### each gene
##### 59494; disease_individual_order; ACC1 ...
##### 59495; disease; ACC...
##### 123824; project_id_tissue_2; ACC_Tumor ...
##### 59460; samples_tissue_type; Tumor, Normal
gene <- list()
for(i in 1:ncol(tumor[,2:59423])){
gene[[i]] <- tumor[,c(1, 59494, 59495, 59460, 123824, 2+i-1)]
}
names(gene) <- colnames(tumor[,2:59423])
enrichment <- list()
#### each geneset
for(i in 1:ncol(tumor[,59498:123823])){
enrichment[[i]] <- tumor[,c(1, 59494, 59495, 59460, 123824, 59498+i-1)]
}
names(enrichment) <- colnames(tumor[,59498:123823])
stats <- c(gene, enrichment)
### dataset with normal
#### each gene
gene_w_normal <- list()
for(i in 1:ncol(violin[,2:59423])){
gene_w_normal[[i]] <- violin[,c(1, 59494, 59495, 59460, 123824, 2+i-1)]
}
names(gene_w_normal) <- colnames(violin[,2:59423])
enrichment_w_normal <- list()
#### each geneset
for(i in 1:ncol(violin[,59498:123823])){
enrichment_w_normal[[i]] <- violin[,c(1, 59494, 59495, 59460, 123824, 59498+i-1)]
}
names(enrichment_w_normal) <- colnames(violin[,59498:123823])
stats_w_normal <- c(gene_w_normal, enrichment_w_normal)
#### stats for all disease
Q50_all <- list()
Q25_all <- list()
Q75_all <- list()
for(i in 1:length(stats)){
Q50_all[i] <- median(stats[[i]][,6])
Q25_all[[i]] <- quantile(stats[[i]][,6], probs = 0.25)
Q75_all[[i]] <- quantile(stats[[i]][,6], probs = 0.75)
}
names(Q50_all) <- names(stats)
names(Q25_all) <- names(stats)
names(Q75_all) <- names(stats)
#### stats for each signature
stats_2 <- list()
for (i in 1:length(stats)) {
df <- stats[[i]]
geneset <- names(df)[6]
##### .data[[geneset]] is dynamic reference in data.frame and tibble, not list.
stats_2[[i]] <- df %>% group_by(disease) %>%
summarise(
mean = mean(.data[[geneset]], na.rm = TRUE),
sd_each = sd(.data[[geneset]], na.rm = TRUE),
Q50_each = quantile(.data[[geneset]], probs = 0.5, na.rm = TRUE),
Q25_each = quantile(.data[[geneset]], probs = 0.25, na.rm = TRUE),
Q75_each = quantile(.data[[geneset]], probs = 0.75, na.rm = TRUE),
min_each = min(.data[[geneset]], na.rm = TRUE),
max_each = max(.data[[geneset]], na.rm = TRUE),
FC_each = quantile(.data[[geneset]], probs = 0.5, na.rm = TRUE) / Q50_all[[i]],
n_each = n(),
.groups = "drop"
) %>% mutate(
color = case_when(
Q50_each <= Q25_all[[i]] ~ "0_low",
Q50_each >= Q75_all[[i]] ~ "2_high",
Q50_each > Q25_all[[i]] & Q50_each < Q75_all[[i]] ~ "1_even"))
}
names(stats_2) <- names(stats)
view(stats_2[[1]])
view(stats[[1]])
view(stats_w_normal[[1]])
#### 2025 11 05
#### color matching
length(stats_w_normal[[1]]$disease)
length(stats_2[[1]]$disease)
color_matching <- list()
for (i in 1:length(stats)) {
color_matching[[i]] <- match(stats_w_normal[[i]]$disease, stats_2[[i]]$disease)
stats_w_normal[[i]]$color <- stats_2[[i]]$color[color_matching[[i]]]
}
table(stats_w_normal[[3]]$color) # Check the code was running normally
#### If log10 scale is better for y-axis, use "scale_y_log10() +" before "theme()"
#### Check the gene or geneset name you need.
gene_geneset_name <- data.frame(target_gene_geneset = names(stats_w_normal))
ウィルコクソンの順位和検定(Mann-WhitneyのU検定)で正常組織と腫瘍組織の遺伝子発現量やエンリッチメントスコアを検定する
せっかく各疾患に対応する正常組織の遺伝子発現量と遺伝子セットのエンリッチメントスコアがあるだから、これを検定しておこうと思う。これは明らかにノンパラメトリックなデータになるわけなので、ウィルコクソンの順位和検定(Mann-WhitneyのU検定)を使って中央値を比較する。
これは各疾患における正常組織と腫瘍組織の遺伝子発現量もしくはエンリッチメントスコアの比較になるので、まずはどんな疾患があるのかを示すデータフレームを作る。リストstats_w_normalから、どの要素でも良い(どの要素;データフレームにも同じだけの症例数が入っているはずなので、どの要素を選んでも同じはず)ので列diseaseの集計をtable()で示し、その結果をデータフレームdiseaseとする。このデータフレームdiseaseは、wilcox.test()で比較する疾患を選ぶときに使用する。
次にウィルコクソンの順位和検定をwilcox.test()を使って行う。やっていることは、リストstats_w_normalの各要素、つまり、ある遺伝子のカウント値もしくはある遺伝子セットのエンリッチメントスコアを、疾患毎に取ってきて、それを列samples_tissue_typeでpivot_wider()で列Normalと列Tumorに広げ、その列をwilcox.test()で比較し、その結果をmutate()を使って列p_valueに入れる。このとき注意すべき点は、すべてのすべての疾患が対応する正常組織のカウント値を持っていない点である。つまり、列NormalがNAの行が沢山あるってことである。この対応として、tryCatch()を使って、もしwilcox.test()でエラーが出たら、列p_valueにNAを入れるようにする。そして、その結果をmutate()に渡し、列p_valueが0.05より低ければ「3_significant」、0.05以上なら「4_not_significant」、それ以外ならNAを列significanceに入れるようにする。それをmutate()にわたし、次は列color_2に以下のように分類を設定する。
| 列project_id_tissue_2の値 | 列colorの値 | 列significanceの値 | 新しく作る列color_2の値 |
|---|---|---|---|
| 文字列「_Tumor」を含む | 0_low | 0_low | |
| 文字列「_Tumor」を含む | 1_even | 1_even | |
| 文字列「_Tumor」を含む | 2_high | 2_high | |
| 文字列「_Normal」を含む | 3_significant | 3_significant | |
| 文字列「_Normal」を含む | 4_not_significant | 4_not_significant | |
| 文字列「_Normal」を含む | NA | 4_not_significant | |
| 上記以外 | 上記以外 | 上記以外 | NA |
それを次はselect()に渡し、列case、列disease_individual_order、列disease、列project_id_tissue_2、列color、color_2、列p_value,significanceを取ってきて、それをリストtempの各要素として入れていく。これをfor()によりリストstats_w_normalの各要素、すなわち各遺伝子と各遺伝子セットで計算していく。
この計算はずいぶんと複雑で時間がかかるので、現在どの要素を計算しているのかがわかるように「Now processing 1 out of 123748 genes」と表示させておいた。すでにメモリをかなり圧迫しており、SWAPを使いまくっているはずなので、計算がいちいち遅くなると思う。これがSWAP Threshingだろうと理解している。コメントによれば、この計算で8時間くらい必要だったらしい。
# code 50
# wilcox.test, normal vs tumor
# use violin
view(stats_w_normal[[1]])
disease <- table(stats_w_normal[[1]]$disease) %>% data.frame()
disease <- disease %>% rename("disease" = "Var1")
disease[1,1]
wilcox <- list()
temp <- list()
start_wilcox <- Sys.time()
# for(x in 1:5){
# for(x in 1:length(stats_w_normal)){
for(x in 1:length(stats_w_normal)){
print(paste0("Now processing ", x, " out of ",length(stats_w_normal)," genes"))
for(y in 1:nrow(disease)){
temp[[y]] <- stats_w_normal[[x]][stats_w_normal[[x]]$disease %in% disease[y,1],] %>% pivot_wider(
names_from = samples_tissue_type,
values_from = names(stats_w_normal)[x]) %>% mutate(
p_value = tryCatch({wilcox.test(Tumor, Normal, alternative = "two.sided")$p.value}, error = function(e) {NA})
) %>% mutate(
significance = case_when(
p_value < 0.05 ~ "3_significant",
p_value >= 0.05 ~ "4_not_significant",
TRUE ~ NA
)
) %>% mutate(
color_2 =case_when(
str_detect(project_id_tissue_2, "_Tumor") == TRUE & color == "0_low" ~ "0_low",
str_detect(project_id_tissue_2, "_Tumor") == TRUE & color == "1_even" ~ "1_even",
str_detect(project_id_tissue_2, "_Tumor") == TRUE & color == "2_high" ~ "2_high",
str_detect(project_id_tissue_2, "_Normal") == TRUE & significance == "3_significant" ~ "3_significant",
str_detect(project_id_tissue_2, "_Normal") == TRUE & significance == "4_not_significant" ~ "4_not_significant",
str_detect(project_id_tissue_2, "_Normal") == TRUE & significance == NA ~ "4_not_significant",
TRUE ~ NA
)
) %>% select(case, disease_individual_order, disease, project_id_tissue_2, color, color_2, p_value,significance)
}
names(temp) <- disease$disease
wilcox[[x]] <- temp
}
names(wilcox) <- names(stats_w_normal)
end_wilcox <- Sys.time()
# at the end of the for() chunk, pivot_longer will be better than select() but several dataset does not have Normal column and therefore pivot_longer() produces error.
duration_wilcox <- end_wilcox - start_wilcox # about 8 hours spent.最終的にはwilcox.test()は遺伝子毎のカウント値が、さらに疾患毎に別れている。なので、ここで疾患毎に各要素に別れているリストwilcoxを縦方向に結合し、各遺伝子もしくは各遺伝子セット毎に全疾患の検定結果を保持したリストwilcox_merge1を作る。これにはreduce()でrbind()を行えば良い。
それを、中央値で階層化を行ったデータを保持したリストstats_w_normalにinner_join()する。これもreduce()でinner_join()を実行すれば出来る。
ここでもかなりの数の要素にrbind()やinner_join()を行うので、print()を使って現時点でどの要素を結合しているのか表示させる。こちらのほうが安心だし、少なくてもコードが動いているのか、止まっているのかわかる。この時点で、おそらくSWAP Thrashingが起こりまくっているので、コードが止まっているように見えるが、そのうちまた動きだす。
# code 51
wilcox_merge1 <- list()
for(i in 1:length(wilcox)){
print(paste0("Now processing ", i, " out of ",length(stats_w_normal)," genes"))
wilcox_merge1[[i]] <- reduce(wilcox[[i]], rbind)
}
names(wilcox_merge1) <- names(stats_w_normal)
# Check size
length(stats_w_normal) # 123748
length(wilcox_merge1) # 123748
stats_w_normal_w_wilcox <- list()
# for(i in 1:5){
# for(i in 1:length(stats_w_normal)){
for(i in 1:length(stats_w_normal)){
print(paste0("Now processing ", i, " out of ",length(stats_w_normal)," genes"))
stats_w_normal_w_wilcox[[i]] <- inner_join(stats_w_normal[[i]], wilcox_merge1[[i]])
}
names(stats_w_normal_w_wilcox) <- names(stats_w_normal)
ここではバイオリンプロットで表示する25パーセンタオル、50パーセンタイル、75パーセンタイルを表示させるためのリストtumor_violinを用意する。これまでのパーセンタイルは、バイオリンプロット自体のある疾患の色を決めるための計算だったが、これはバイオリンプロットに引くパーセンタイルの水平線を計算するための計算である。このためにリストstats_w_normal_w_wilcoxの各要素(すなわち各遺伝子もしくは各遺伝子セットのデータフレーム)から、列project_id_tissue_2に「_Tumor」という文字列を含む例を取ってきて、リストtumor_violinを作成すれば良い。ここにもどの要素を処理しているのかわかる用にprint()でコメントを入れる。
# code 52
tumor_violin <- list()
for(i in 1:length(stats_w_normal_w_wilcox)){
print(paste0("Now processing ", i, " out of ",length(stats_w_normal)," genes"))
tumor_violin[[i]] <- stats_w_normal_w_wilcox[[i]] %>% filter(str_detect(project_id_tissue_2, "_Tumor") == TRUE)
}
names(tumor_violin) <- names(stats_w_normal)
バイオリンプロットを表示する
最初に、自分が見たい遺伝子もしくは遺伝子セットに当たるデータフレームを、リストstats_w_normal_w_wilcoxから取ってきて、それをデータフレームgene_geneset_dfに入れる。ここでは、遺伝子セットHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaを見たいと思う。また、データフレームを変数として参照したいので、そのデータフレーム名、すなわち、遺伝子名もしくは遺伝子セット名を取得して長さ1のベクトルgene_geneset_showとする。それを使って、対応するデータフレームをリストtumor_violinから取得してきて、データフレームgene_geneset_df_tumorとする。そのデータフレームを使ってバイオリンプロット上に25、50、75パーセンタイルの水平線を引く。
そして、ようやっとバイオリンプロットである。ggplot2では最初にggplot()で使用するデータを設定しなければならない。ここでは、データフレームgene_geneset_df、横軸には列project_id_tissue_2、縦軸には、ここが長さ1のベクトルgene_geneset_showに入っている列を使用する。縦軸が変数になるので、.data[[変数]]を使用する。tidyverseパッケージでは、この例のデータフレームgene_geneset_dfのように、既に参照されている場合にこの.dataを使用することが出来る(と個人的には理解している)。バイオリンプロットの描画にはgeom_violin()を使用する。パーセンタイルの水平線にはgeom_hline()を使う。このとき、水平線としてデータフレームgene_geneset_df_tumorの長さ1のベクトルgene_geneset_showに相当する列の25パーセンタイル、50パーセンタイル、75パーセンタイルをquantile()で求め、それをgeom_hline()の引数yinterceptに入れる。それぞれの色、すなわち青、黒、赤を引数colorに入れれば、25パーセンタイル,50パーセンタイル、75パーセンタイルをそれらの色で引くことが出来る。バイオリンプロットの色は、scale_fill_manual()に入れる。以下に色とその色の意味をまとめる。これを流すとバイオリンプロットが表示される。
| 組織 | 列color_2の値 | 色 | 意味 |
|---|---|---|---|
| 腫瘍 | 0_low | lightblue | その腫瘍組織の50パーセンタイル(中央値)が、疾患全体の25パーセンタイル以下。 |
| 腫瘍 | 1_even | gray | その腫瘍組織の50パーセンタイル(中央値)が、疾患全体の25パーセンタイルより高く、75パーセンタイルより低い。 |
| 腫瘍 | 2_high | red | その腫瘍組織の50パーセンタイル(中央値)が、疾患全体の75パーセンタイル以上 |
| 正常 | 3_significant | lightgreen | 正常組織と腫瘍組織のカウント値に統計的有意差あり |
| 正常 | 4_not_significant | gray | 正常組織と腫瘍組織のカウント値に統計的有意差なし |
これで、このcode 52の最初の、gene_geneset_df <- stats_w_normal_w_wilcox[[“HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva”]]、のところで見たい遺伝子もしくは遺伝子セットを指定してこのコードチャンクを流すだけで、バイオリンプロットが表示出来る。
もしそれを画像として保存したいときにはこの例の用にtiff()や、これはggplot2なので、そのままggsave()を使えば良い。個人的には細かく設定出来るtiff()が好きである。あと、ggsave()にはあまり慣れてないし。
# code 53
### -------------- run below to show violin plot v.4----------------
### Run following code to show violin plot of the gene or geneset of interest.
#### Specify gene or geneset to be shown.
gene_geneset_df <- stats_w_normal_w_wilcox[["HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva"]] # HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva # CSNK2A1
gene_geneset_show <- names(gene_geneset_df)[6]
gene_geneset_df_tumor <- tumor_violin[[gene_geneset_show]]
#### check dataset
view(stats_w_normal_w_wilcox[[gene_geneset_show]])
table(stats_w_normal_w_wilcox[[gene_geneset_show]]$color_2)
table(stats_w_normal_w_wilcox[[gene_geneset_show]]$project_id_tissue_2,stats_w_normal_w_wilcox[[gene_geneset_show]]$color_2)
#### show violin plot
ggplot(gene_geneset_df, aes(x = project_id_tissue_2, y = .data[[gene_geneset_show]], fill = color_2)) +
geom_violin(width = 2, trim = FALSE) +
geom_hline(yintercept = quantile(gene_geneset_df_tumor[[gene_geneset_show]], probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "red", "lightgreen", "gray")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
#### color of violin plot;
## 0_low; "lightblue"
## 1_even; "gray"
## 2_high; "red"
## 3_significant; "lightgreen"
## 4_not_significant; "gray"
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/",gene_geneset_show,"", ".tiff", sep = "")
tiff(filename = path,
width = 2500, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
ggplot(gene_geneset_df, aes(x = project_id_tissue_2, y = .data[[gene_geneset_show]], fill = color_2)) +
geom_violin(width = 2, trim = FALSE) +
geom_hline(yintercept = quantile(gene_geneset_df_tumor[[gene_geneset_show]], probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "red", "lightgreen", "gray")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
dev.off()
以下の画像がcode 52を流して得られたバイオリンプロットである。見方としては、ある疾患が、正常組織が緑(正常組織と腫瘍組織におけるある遺伝子のカウント値の中央値もしくはある遺伝子セットのエンリッチメントスコアの中央値に統計的有意差あり)、かつ、腫瘍組織が赤(遺伝子のカウント値もしくは遺伝子セットのエンリッチメントスコアの中央値が、全疾患の中央値よりの1.5倍以上)で示されている場合に、選んだ遺伝子の発現量もしくは遺伝子セットのエンリッチメントスコアに何らかの違いがありそうということになる。この例で言えば、HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva、すなわち、GSVAでエンリッチされてきたIFNgのシグナリングは、おそらくHNSC(頭頸部扁平上皮癌)、LUAD(肺腺癌)で有意に高そいかもしれないことがわかる。また、LUSC(肺扁平上皮癌)では、IFNgのシグナリングは、全疾患と比べるとそうでもないが、正常組織と比較すると有意に低くなっている可能性があるとわかる。注意点の一つ目は正常組織の例数が少ないことである。以下の画像の例では、CHOL_NormalやREAD_Normalの症例が特に少ないことがわかる。例数が少ないため、横方向に長い分布に見えるためだ。例数が多い場合、分布が広いので縦長になる。例数も少ないということは、信頼性がちょっと足りないということでもあると思う。また、正常組織のカウントが取られていない疾患も多い。よって、TCGAの解析では無理に正常組織を含めて解析する必要は内容に思う。
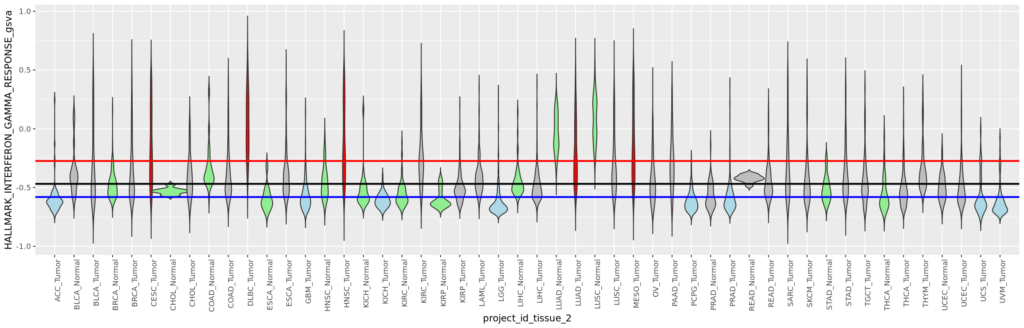
カプランマイヤー法による生存率の解析
以降でカプランマイヤー法による生存期間の解析を行うが、まずは各遺伝子のカウント値もしくは各遺伝子セットのエンリッチメントスコアを使って患者を階層化し、次にカプランマイヤー法で生存率曲線(カプランマイヤー生存率、カプランマイヤー推定値、カプランマイヤー曲線とかと言うようだ。ここでは生存率曲線という。この生存率曲線という呼び名は、新版Cox比例ハザードモデル、ISBN978-4-254-12884-0でも使用されているので、ここでもそれを使うことにする)を作成する。最後に階層間のログランク検定を行い、その結果をまとめてファイルに出力しておこうと思う。
打ち切りの設定
カプランマイヤー法による生存期間の解析では、正常組織のカウント値は必要ない。ここではcode 49で作成した、データフレームviolinから、列samples_tissue_typeの値が「Tumor」の行を抽出して作成したデータフレームtumorを使用する。
カプランマイヤー曲線を描くためには、打ち切りを設定する必要がある。追跡が死亡や何らかの理由でフォローアップを止まってしまった患者を打ち切りとして1、生存して観察が続いている例を0として計算する。以下の表の用に分類をした。この分類では列の値を使いたいので、if()、else_if()、else()を用いた。もしかしたらcase_when()でも出来るかもしれないが、それは試していない。
| 列demographic_vital_status | 列demographic_days_to_death | 列diagnoses_days_to_last_follow_up | 新しく作る列diagnoses_days_to_last_follow_up_2 |
|---|---|---|---|
| Alive | NA | 列diagnoses_days_to_last_follow_upの値 | |
| Dead | NA | 列demographic_days_to_deathの値 | |
| Dead | NA | 列diagnoses_days_to_last_follow_upの値 | |
| その他 | その他 | その他 | 列diagnoses_days_to_last_follow_upの値 |
その分類が出来たら、新しく作成した列diagnoses_days_to_last_follow_up_2が0よりも大きい症例を取ってきて、それをデータフレームtumor_2とし、この列の名前をシンプルにDayに変えておいた。
ここで少し悩んだんだが、打ち切り(Censor)は、教科書(生存時間解析、5ページ、表1.1、ISBN978-4-254-12861-1)に従って設定した。生存率曲線の横軸は、1825日(365日x5年=1825日)とした。後述するが、これは観察されている全期間における全生存期間になる。
TCGAのデータにおける打ち切りの設定はけっこう迷うところである。TCGAのデータは、場合によっては5000日を超えるような症例もあり、そんなに長いのは一般的では無いと思う。また、有限期間、例えば5年(365日x5年=1825日)にしないと、打ち切り(Censor)を上手く設定出来ないように思う。なので、評価期間を1825日として、それより長く生きた症例をCensorを0(生存)、Aliveではあるが列Dayが1825日以前ならばCersorを1(打ち切り)、Deadではあるがその期間が1825日以降ならばCensorを0、Deadでその期間が1825日以前ならばCensorを1(死亡)として計算するのもありだと思う。教科書(生存時間解析、5ページ、表1.1、ISBN978-4-254-12861-1)に倣って打ち切りを設定すると、それは全生存期間(Overall Survival;OS)、しかも、観察されている全期間における全生存期間になると思う。ただし、これだと打ち切りが多すぎで信頼できないと言われる可能性がある。その場合は、5年生存率にしたらよい。打ち切りのほとんどない生存率曲線が得られる。こちらの方が適切かもしれない。
打ち切りは、再発したり、PD(Progressive Disease;治療後のフォローアップのときにCTで評価される腫瘍体積や血液検査での腫瘍マーカーの値が治療開始時に比べて悪化したりした場合)だった場合にも当てはまる可能性があるので、目的に応じて適切に設定しなけれならない。この場合の生存率は無病生存率(無病生存期間)(Disease Free Survival;DFS)とか無憎悪生存率(無憎悪生存期間)Progression Free Survival;PFS)とかである。OSとDFSの両方を示す論文も多い。
# code 54
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster.
tumor <- read_tsv("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/tumor.tsv")
# check case with dead or alive.
table(tumor$demographic_vital_status)
# Alive Dead Not Reported Unknown
# 7144 3098 1 2
# case with "Not reported" will be removed.
tumor_2 <- tumor %>% filter(demographic_vital_status == "Alive" | demographic_vital_status == "Dead")
# prepare empty column.
tumor_2$diagnoses_days_to_last_follow_up_2 <- c(rep(NA, nrow(tumor_2))) %>% as.numeric()
for(i in 1:nrow(tumor_2)){
if(tumor_2$demographic_vital_status[i] == "Alive" & is.na(tumor_2$demographic_days_to_death[i]) == TRUE){
tumor_2$diagnoses_days_to_last_follow_up_2[i] <- tumor_2$diagnoses_days_to_last_follow_up[i]}
else if(tumor_2$demographic_vital_status[i] == "Dead"& is.na(tumor_2$diagnoses_days_to_last_follow_up[i]) == TRUE){
tumor_2$diagnoses_days_to_last_follow_up_2[i] <- tumor_2$demographic_days_to_death[i]}
else if(tumor_2$demographic_vital_status[i] == "Dead"& is.na(tumor_2$demographic_days_to_death[i]) == TRUE){
tumor_2$diagnoses_days_to_last_follow_up_2[i] <- tumor_2$diagnoses_days_to_last_follow_up[i]}
else{tumor_2$diagnoses_days_to_last_follow_up_2[i] <- tumor_2$diagnoses_days_to_last_follow_up[i]}
}
tumor_2 <- tumor_2 %>% filter(diagnoses_days_to_last_follow_up_2 > 0)
tumor_2 <- tumor_2 %>% rename("Day" = "diagnoses_days_to_last_follow_up_2")
# In case of the all observed periods
tumor_2 <- tumor_2 %>% mutate(
survival = case_when(
demographic_vital_status == "Alive" ~ 0,
demographic_vital_status == "Dead" ~ 1
)
)
# # 5 years survival analysis. It can be used for survival analysis
# tumor_2 <- tumor_2 %>% mutate(
# survival = case_when(
# demographic_vital_status == "Alive" & Day >= 1825 ~ 0,
# demographic_vital_status == "Alive" & Day < 1825 ~ 1,
# demographic_vital_status == "Dead" & Day >= 1825 ~ 0,
# demographic_vital_status == "Dead" & Day < 1825 ~ 1,
# )
# )ちょっと話が反れるが、自分のコンピューターではこのあたりでおそらくメモリ圧迫によるSWAP Threshingが酷いことになってきたので、コンピューターやソフトがいつクラッシュしてしまっても大丈夫なように、以降に重要なデータフレームはwrite_tsv()で出力しておく。
# code 55
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
write_tsv(tumor_2, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/tumor_2.tsv")次に、データフレームtumor_2から、1列目(列case)、59494列目(列disease_individual_order)、59495列目(列disease)、59467列目(列project_id)、123825列目(列Day)、123826列目(列survival)、2列目から59423列目(遺伝子)、59498列目から123823列目(遺伝子セット)を選んできて、新しくデータフレームtumor_3を作成する。
# code 56
# just look at the columns.
colnames(tumor_2[,59420:59500])
# column 1; case; case1, case2...
# column 59494; disease_individual_order; ACC1, ACC2...
# column 59495; disease; ACC...
# column 59467; project_id; TCGA-ACC...
# column 123825; Day; see the code chunk just above.
# column 123826; survival; see the code chunk just above.
# column 2:59423; gene expression
# column 59498:123823; results of enrichment
# In addition to "case", "disease_individual_order", "disease", "project_id", Day","survival", choose required columns.
tumor_3 <- tumor_2[,c(1, 59494, 59495, 59467, 123825, 123826, 2:59423, 59498:123823)]このデータフレームtumor_3もwrite_tsv()で出力しておく。
# code 57
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
write_tsv(tumor_3, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/tumor_3.tsv")以降はデータフレームtumor_3を使っていくので、データフレームtumorとデータフレームtumor_2はremove()で削除してしまってもいいし、一度Rを閉じて、また立ち上げ直して、tumor_3.tsvをfread()やread_tsv()などで改めて読み込んでも良いと思う。というか、自分はそうした。
# code 58
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
tumor_2 <- read_tsv("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/tumor_2.tsv")
tumor_3 <- read_tsv("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/tumor_3.tsv")ここでcode 49あたりのバイオリンプロットのためのリストを作成したときと同様に、各遺伝子もしくは各遺伝子セット要素とするリストanalysis_survivalを作成する。進捗状況を確認するためにprint()で現時点で処理されている要素を表示させておく。
# code 59
# column 1; case; case1, case2...
# column 59494; disease_individual_order; ACC1, ACC2...
# column 59495; disease; ACC...
# column 59467; project_id; TCGA-ACC...
# column 123825; Day; see the code chunk just above.
# column 123826; survival; see the code chunk just above.
# column 2:59423; gene expression
# column 59498:123823; results of enrichment
analysis_survival <- list()
# view(analysis_3[,c(1:6,7)]) # check them
for(i in 7:ncol(tumor_3)){
print(paste0("Now processing ", i-6, " out of ", ncol(tumor_3)-6, " genes"))
analysis_survival[[i-6]] <- tumor_3[,c(1:6,i)] %>% data.frame()}
names(analysis_survival) <- colnames(tumor_3[,7:ncol(tumor_3)])ここでも、バイオリンプロットでの分類と同様に、各疾患(列disease)における各遺伝子のカウント値の25パーセンタイル、50パーセンタイル、75パーセンタイルを計算し、各症例におけるある遺伝子のカウント値がその疾患のパーセンタイルと比較してどのくらい高いのか、それとも低いのかで各々の症例を分類する。ここでもバイオリンプロットのときと同様に、変数を用いた動的な列とデータフレームの参照方法を使用する(というか、これを使わないとエラーが出てしまっていた)。まずは各遺伝子のカウント値を入れた列である、リストanalysis_survivalの各要素の7列目のから、遺伝子もしくは遺伝子セット名を取ってきてベクトルgene_name_survivalとする。また、リストanalysis_survivalの各要素をデータフレームtemp_survival_dfに入れる。このデータフレームtemp_survival_dfには、疾患毎の生存期間、打ち切りの値(列survival)などが入っている。そのデータフレームtemp_survival_dfの列diseaseでgroup_by()し、疾患毎の25パーセンタイル(列Q25)、50パーセンタイル(列Q50)、75パーセンタイル(列Q75)、疾患の数(列n)を求め、新しくデータフレームtemp_survival_quantileを作成する。次に、データフレームtemp_survival_dfの列diseaseとデータフレームtemp_survival_quantileの列diseaseが一致しているかどうかをmatch()で比較し、データフレームtemp_survival_quantileの列Q25、列Q50、列Q75の値を、データフレームtemp_survival_dfに反映させる。次に、その列Q25、列Q50、列Q75の値を以下のように分類する。
| analysis_survival[[i]][[gene_name_survival]](データフレームanalysis_survival[[i]]の変数gene_name_survivalで示される列の値 | 新しく作る列layer_2 | 新しく作る列layer_3 |
|---|---|---|
| データフレームtemp_survival_dfの列Q50以上 | low | |
| データフレームtemp_survival_dfの列Q50より低い | high | |
| データフレームtemp_survival_dfの列Q75以上 | low | |
| データフレームtemp_survival_dfの列Q25より高くQ75より低い | mid | |
| データフレームtemp_survival_dfの列Q25以下 | high |
この列layer_2と列layer_3は生存率曲線の階層に使用する。また、この階層がグラフ毎に順番が変わると非常に面倒なので、factor()でその順序を定めておく。
# code 60
# USE for(), DO NOT USE foreach(). foreach() spent huge amount of memory, which was more than 1 TB, and it took very long time to store the data for the execution.
for(i in 1:length(analysis_survival)){
print(paste0("Now processing ", i, " out of ", length(analysis_survival), " genes"))
# IMPORTANT; Need to use ".data[[]]" not "temp_survival_df"
gene_name_survival <- names(analysis_survival[[i]][7])
temp_survival_df <- analysis_survival[[i]] %>% data.frame()
temp_survival_quantile <- temp_survival_df %>% group_by(disease) %>%
summarise(Q50 = quantile(.data[[gene_name_survival]], probs = 0.50, na.rm = TRUE),
Q25 = quantile(.data[[gene_name_survival]], probs = 0.25, na.rm = TRUE),
Q75 = quantile(.data[[gene_name_survival]], probs = 0.75, na.rm = TRUE),
n = n())
## color matching
# color_matching <- match(analysis$disease, stats_EMT$disease)
# analysis$color <- stats_EMT$color[color_matching]
disease_matching <- match(temp_survival_df$disease, temp_survival_quantile$disease)
temp_survival_df$Q50 <- temp_survival_quantile$Q50[disease_matching]
temp_survival_df$Q25 <- temp_survival_quantile$Q25[disease_matching]
temp_survival_df$Q75 <- temp_survival_quantile$Q75[disease_matching]
analysis_survival[[i]] <- analysis_survival[[i]] %>%
mutate(
Q25 = temp_survival_df$Q25,
Q50 = temp_survival_df$Q50,
Q75 = temp_survival_df$Q75,
layer_2 = case_when(analysis_survival[[i]][[gene_name_survival]] >= temp_survival_df$Q50 ~ "high",
analysis_survival[[i]][[gene_name_survival]] < temp_survival_df$Q50 ~ "low"),
layer_3 = case_when(analysis_survival[[i]][[gene_name_survival]] >= temp_survival_df$Q75 ~ "high",
analysis_survival[[i]][[gene_name_survival]] > temp_survival_df$Q25 & analysis_survival[[i]][[gene_name_survival]] < temp_survival_df$Q75 ~ "mid",
analysis_survival[[i]][[gene_name_survival]] <= temp_survival_df$Q25 ~ "low")
)
# set order for layer_2
analysis_survival[[i]]$layer_2 <-
factor(analysis_survival[[i]]$layer_2,
levels = c("high", "low"),
labels = c("high", "low"),
exclude = NA, ordered = is.ordered(analysis_survival[[i]]$layer_2), nmax = NA)
# set order for layer_3
analysis_survival[[i]]$layer_3 <-
factor(analysis_survival[[i]]$layer_3,
levels = c("high", "mid", "low"),
labels = c("high", "mid", "low"),
exclude = NA, ordered = is.ordered(analysis_survival[[i]]$layer_3), nmax = NA)
}
# check data
view(analysis_survival[[2]])
table(analysis_survival[[1]]$layer_2)
table(analysis_survival[[1]]$layer_3)生存率曲線の表示
生存率の表示も、バイオリンプロットと同様に遺伝子名もしくは遺伝子セット名を一回だけ入力するだけで、後はコードを流せば目的の図が出来上がるようにする。リストanalysis_survivalから遺伝子名もしくは遺伝子セット名を取ってきて、列名をgene_genesetとし、それをデータフレームgene_name_survival_2とし、これを使って興味のある遺伝子名もしくは遺伝子セット名を探すことにする。そして、ここではHALLMARK_INTERFERON_GAMMA_RESPONSE_gsvaの生存率を比較したいと思う。「HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva」を長さ1のベクトルgene_geneset_interestに入れる。また、それぞれの疾患の生存率を描画するときに疾患名を参照する必要もあるので、リストanalysis_survivalに入っている要素(のデータフレーム。これは何でも良い)から列diesaseを取ってきて、それをunique()に渡し重複を除去し、それをデータフレームdisease_nameとした。このデータフレームdisease_nameの列名をdiseaseにする。
# code 61
gene_name_survival_2 <- data.frame(gene_geneset = names(analysis_survival)) # for looking up gene or geneset of interest
view(gene_name_survival_2) # look for gene or geneset of interest
gene_geneset_interest <- "HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva" # put gene or geneset of interest for survival curve
# HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva
# CSNK2A1
#
disease_name <- analysis_survival[[1]]$disease %>% unique() %>% data.frame() # any number is OK.
colnames(disease_name) = "disease"リストanalysis_survivalから、長さ1のベクトルgene_geneset_interestに入っている文字列と一致する遺伝子もしくは遺伝子セット(ここではHALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)のデータフレームから、データフレームdiesase_nameの列diseaseで示される疾患を取ってきて、それをリストinterest_gene_geneset_survival_1に入れる。ここでは最初に1から18行目の疾患をfor()でリストinterest_gene_geneset_survival_1に入れている。本当は33個の生存率曲線を一気に表示したいのだが、そうすると多くの画像を一つの図に入れることになり、各グラフが小さく潰れて読めなくなってしまうためである。なので、まずは1から18行目の疾患を表示させる。次に、このリストinterest_gene_geneset_survival_1の各要素を、生存期間として列Day、打ち切りの値として列survival、階層として列layer_2をSurv()に入れ、生存期間や打ち切りの値を設定し、それを使ってsurvfit()で統計量を計算する。その結果をリストfit_status_1の各要素に入れていく。このリストfit_status_1の各要素をggsurvplot()に入れて、生存率曲線のグラフを作成し、それをリストsurvival_curve_status_1の各要素に入れていく。そして、リストsurvival_curve_status_1の各要素にlapply()でplot()を適用し、それをリストsurvival_curve_status_1_graphに入れていく。最後にgrid.arrange()でパネル化する。画像の保存にはtiff()を使う。
# code 62
# 1 - 18 project.
# prepare list
interest_gene_geneset_survival_1 <- list()
for(i in 1:18){
interest_gene_geneset_survival_1[[i]] <- analysis_survival[[gene_geneset_interest[1]]] %>% filter(disease == disease_name$disease[i])
} # If length(disease_name) was used, the all 33 project were put in the list. 18 means only 18 projects were put in the list.
names(interest_gene_geneset_survival_1) <- disease_name$disease[1:18]
fit_status_1 <- list() # 1 - 15
survival_curve_status_1 <- list() # 1 - 15
length(interest_gene_geneset_survival_1) # 18 diseases
for(i in 1:length(interest_gene_geneset_survival_1)){
fit_status_1[[i]] <- survfit(Surv(Day, survival) ~ layer_2, data = interest_gene_geneset_survival_1[[i]])
survival_curve_status_1[[i]] <- ggsurvplot(
fit_status_1[[i]],
data = interest_gene_geneset_survival_1[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6),
font.legend = c(6),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i])
} # legend = c("right", "left", "top", "bottom", "none") can show which color is which layer.
# red; high
# green; low
survival_curve_status_1_graph <- lapply(survival_curve_status_1, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_1_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/",gene_geneset_interest,"_2_layer_1",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_1_graph, ncol = 6)
dev.off()以下が出力される生存率曲線である。
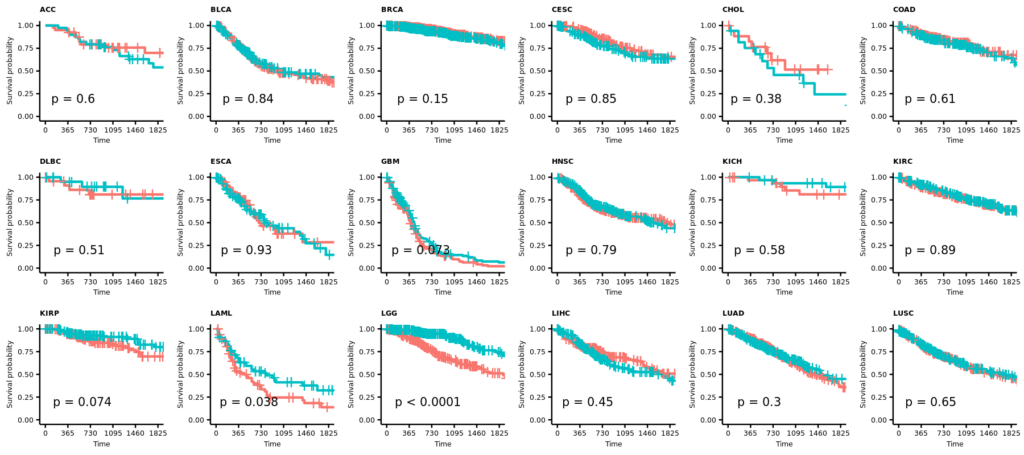
以下は19から33行目の疾患に関するlayer_2の階層での生存率である。
# code 63
# 19 - 33 project.
# prepare list
interest_gene_geneset_survival_2 <- list()
for(i in 19:33){
interest_gene_geneset_survival_2[[i-18]] <- analysis_survival[[gene_geneset_interest[1]]] %>% filter(disease == disease_name$disease[i])
} # If length(disease_name) was used, the all 33 project were put in the list. 18 means only 18 projects were put in the list.
names(interest_gene_geneset_survival_2) <- disease_name$disease[19:33]
fit_status_2 <- list() # 1 - 15
survival_curve_status_2 <- list() # 1 - 15
length(interest_gene_geneset_survival_2) # 15 diseases
for(i in 1:length(interest_gene_geneset_survival_2)){
fit_status_2[[i]] <- survfit(Surv(Day, survival) ~ layer_2, data = interest_gene_geneset_survival_2[[i]])
survival_curve_status_2[[i]] <- ggsurvplot(
fit_status_2[[i]],
data = interest_gene_geneset_survival_2[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "bold", "black"),
font.y = c(6, "bold", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i+18])
} # legend = c("right", "left", "top", "bottom", "none") can show which color is which layer.
# red; high
# green; low
survival_curve_status_2_graph <- lapply(survival_curve_status_2, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_2_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/",gene_geneset_interest,"_2_layer_2",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_2_graph, ncol = 6)
dev.off()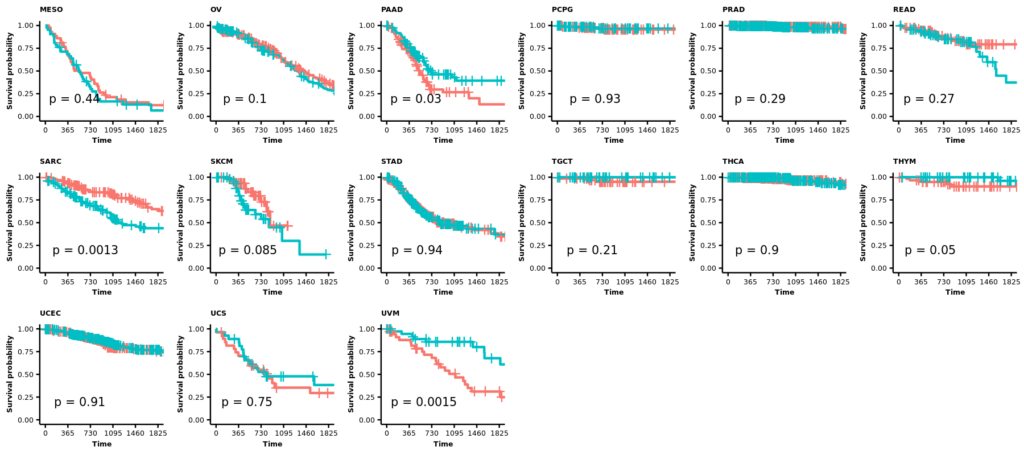
以下は1から18行目の疾患に関するlayer_3の階層での生存率である。
# code 64
# 1 - 18 project.
fit_status_3 <- list() # 1 - 15
survival_curve_status_3 <- list() # 1 - 15
length(interest_gene_geneset_survival_1) # 18 diseases
for(i in 1:length(interest_gene_geneset_survival_1)){
fit_status_3[[i]] <- survfit(Surv(Day, survival) ~ layer_3, data = interest_gene_geneset_survival_1[[i]])
survival_curve_status_3[[i]] <- ggsurvplot(
fit_status_3[[i]],
data = interest_gene_geneset_survival_1[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i])
}# legend = "bottom") can show which color is which layer.
# red; high
# green; mid
# blue; low
survival_curve_status_3_graph <- lapply(survival_curve_status_3, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_3_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/",gene_geneset_interest,"_3_layer_1",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_3_graph, ncol = 6)
dev.off()
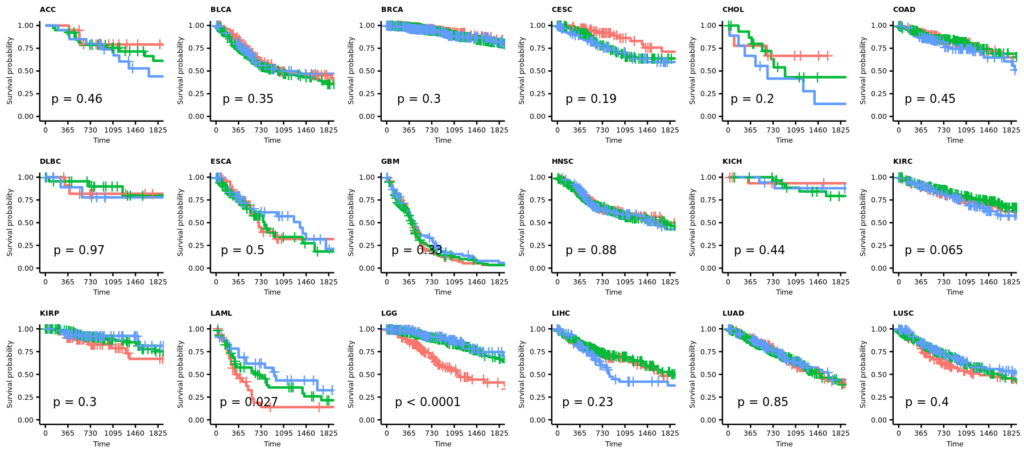
p値はログランク検定の結果。
以下は19から33行目の疾患に関するlayer_3の階層での生存率である。
# code 65
# 19 - 33 project.
fit_status_4 <- list() # 1 - 15
survival_curve_status_4 <- list() # 1 - 15
length(interest_gene_geneset_survival_2) # 15 diseases
for(i in 1:length(interest_gene_geneset_survival_2)){
fit_status_4[[i]] <- survfit(Surv(Day, survival) ~ layer_3, data = interest_gene_geneset_survival_2[[i]])
survival_curve_status_4[[i]] <- ggsurvplot(
fit_status_4[[i]],
data = interest_gene_geneset_survival_2[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i+18])
} # legend = "bottom") can show which color is which layer.
# red; high
# green; mid
# blue; low
survival_curve_status_4_graph <- lapply(survival_curve_status_4, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_4_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/",gene_geneset_interest,"_3_layer_2",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_4_graph, ncol = 6)
dev.off()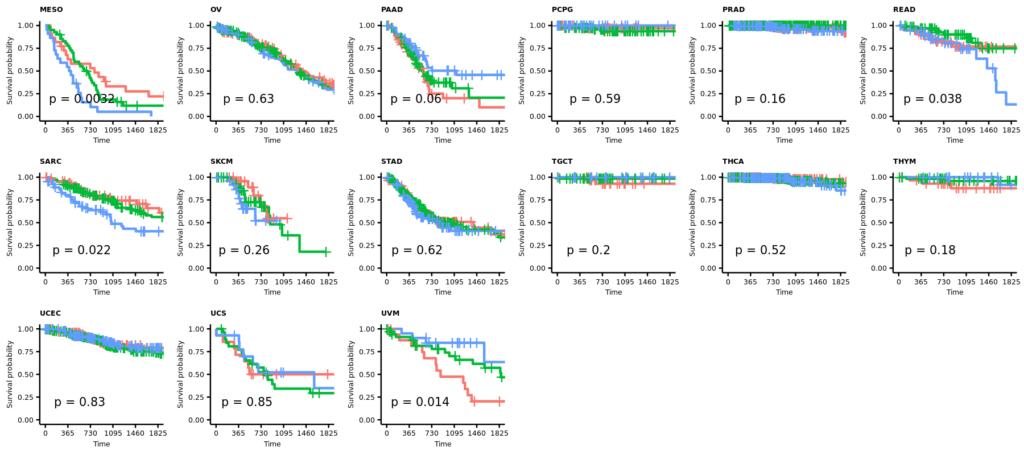
ログランク検定で階層間の統計的有意差を求める
生存率の階層間における統計的有意な違いをログランク検定で求める。まずは、計算に使用するデータフレームをまとめたリストを作成する。また、現時点でどの要素を計算しているのかがわかるように、print()で計算中の要素を表示する。計算する数が多く時間がかかるので、これがわかると精神的に楽だし、ある意味ではトラブルシューティングになる。リストanalysis_survivalの各要素、各遺伝子のカウント値、もしくは各遺伝子セットのエンリッチメントスコアを収めたデータフレームから、列diseaseの値を抜き出してきて、リストtemp_logrank_1に入れる。それを、さらにリストlogrankに入れて行く。このリストlogrankには、まず各遺伝子名もしくは各遺伝子セット名を入れ、そして、その中の要素には列diseaseの値(ACCとかBRCAとかの33個の疾患の分類)毎の生存期間(列Day)と打ち切りの値(列survival)がネストして入っている。
# code 66
# 2025 10 11
# prepare dataset
temp_logrank_1 <- list()
logrank <- list()
for(x in 1:length(analysis_survival)){
print(paste0("Now processing ", x, " out of ", length(analysis_survival), " genes"))
for(y in 1:length(disease_name$disease)){
temp_logrank_1[[y]] <- analysis_survival[[x]] %>% data.frame() %>% filter(disease == disease_name$disease[y])
}
names(temp_logrank_1) <- disease_name$disease
logrank[[x]] <- temp_logrank_1
}
names(logrank) <- names(analysis_survival)計算結果を入れるための空のデータフレームtemp_dataframeを用意する。これは列disease(ACCとかBRCAとか、33個の疾患。すなわち33行)、列layer_2_pval(最初の時点ではNAが33行)、列layer_3_pval(最初の時点ではNAが33行)から成る。
リストlogrankの中にネストされた疾患の生存期間(列Day)と打ち切りの値(列survival)を使って、survdiff()でログランク検定を行い、結果を列layer_2_pvalもしくは列layer_3_pvalに入れていく。これをリストlogrankの全部の要素に対してfor()を使って行っていき、各要素をリストlogrank_pvalに入れる。ここでも、やはりNAを含む疾患に当たるとエラーになってしまうので、その場合はtryCatch()で処理する。このリストlogrank_pvalは、リストlogrankと同じように、まずは遺伝子名もしくは遺伝子セット名で、その下のそうには疾患毎のリストがネストされた構造となっている。
# code 67
# calculate log-rank for all disease over all gene and geneset
temp_dataframe <- data.frame(disease = disease_name$disease, layer_2_pval=rep(NA, 33), layer_3_pval=rep(NA, 33))
logrank_pval <- list()
# if analysis_survival is removed because of too big, use length_analysis_survival and names_analysis_survival
length_analysis_survival <- length(analysis_survival)
names_analysis_survival <- names(analysis_survival)
for(x in 1:length(analysis_survival)){
print(paste0("Now processing ", x, " out of ", length(analysis_survival), " genes"))
for(y in 1:length(disease_name$disease)){
temp_df <- logrank[[x]][[y]] %>% data.frame()
temp_dataframe$layer_2_pval[y] <- tryCatch({survdiff(Surv(Day, survival) ~ layer_2, data = temp_df)$p}, error = function(e) {NA})
temp_dataframe$layer_3_pval[y] <- tryCatch({survdiff(Surv(Day, survival) ~ layer_3, data = temp_df)$p}, error = function(e) {NA})
}
logrank_pval[[x]] <- temp_dataframe
}
names(logrank_pval) <- names(analysis_survival)同じような計算方法で、列layer_2_pvalと列layer_3_pvalの値に対して、FDR(False Discovery Rate)の補正を行い、それぞれ列layer_2_pval_FDR、列layer_3_pval_FDRに入れる。ここでもtryCatch()でエラー処理を入れておく。解析の目的が「この33個の疾患の中から、関心のある遺伝子の発現量もしくは各遺伝子セットのエンリッチメントスコアが深く関わりそうな疾患を拾う」という、他グループとの比較と考えることも出来るので、FDRの補正はやっておいても良いのでは無いかと思う。その一方で、結局独立して比較しているのだから、別にやる必要も無いような気もする。このあたりは今後しっかり勉強しておきたいところである。無難なのは、FDR補正で疾患を選んでくることだと思う。最後にreduce()とbind_rows()(rbind()でも良い)でリストを縦方向に結合し、データフレームlogrank_pval_dfを作成する。
FDRの補正後のp値が0.05という場合、「同時に沢山の検定をして、そこから選んできたもののうち5%は間違ってるかも」という意味と理解している。
# code 68
# FDR correction and name the each column
for(i in 1:length(logrank_pval)){
print(paste0("Now processing ", i, " out of ", length(logrank_pval), " genes"))
logrank_pval[[i]] <- logrank_pval[[i]] %>% mutate(
layer_2_pval_FDR = tryCatch({p.adjust(layer_2_pval, method = "fdr")}, error = function(e) {NA}),
layer_3_pval_FDR = tryCatch({p.adjust(layer_3_pval, method = "fdr")}, error = function(e) {NA}),
gene_geneset = names(logrank_pval[i])
)
}
# get the list together to a data frame
logrank_pval_df <- reduce(logrank_pval, bind_rows) %>% data.frame()ここで、生存期間に統計的な有意差があった疾患のみを拾って来て、どの遺伝子が患者の生存率に影響を当たるのかをまとめて見ることにする。やったことは、データフレームlogrank_pval_dfから列diseaseの値、すなわちACCやBRCA等の33個の疾患をtable()で集計し、ACCやBRCA等の疾患名が入っている列Var1をデータフレームdisease_interestに入れる。ここはそれぞれの疾患名が33個入っているはらば、何でも良いと思う。次にデータフレームlogrank_pval_dfの列diseaseがデータフレームdisease_interestの各要素、すなわち33個の疾患のうちの一つの場合、かつ列layer_2_pval_FDRが0.05より小さい疾患を拾って来てリストdisease_significant_layer_2の各要素に入れていく。これを列layer_3_pval_FDRに対しても行い、リストdisease_significant_layer_3の各要素に入れる。この2つの要素をリストdisease_significantに入れる。
これで、各遺伝子のカウント値もしくは各遺伝子セットのエンリッチメントスコアで階層化した場合の各症例の生存期間に影響を与えそうな遺伝子もしくは遺伝子セットを集めてくることが出来る。これを使うことで、新しい治療標的を見つけることが出来るかもしれない。また、ここの生存率を見ている場合はFDRの補正は不要かもしれないが、こういった多くの物を選んできていいる場合はFDRの補正は必要であるように思う。
# code 69
# example; which genes and geneset are closely associated the survival of PAAD patients??
# PAAD_significant <- logrank_pval_df %>% filter(disease == "PAAD" & (layer_2_pval_FDR < 0.05))
disease_interest <- data.frame(table(logrank_pval_df$disease))$Var1
disease_significant_layer_2 <- list()
for(i in 1:length(disease_interest)){
disease_significant_layer_2[[i]] <- logrank_pval_df %>% filter(disease == disease_interest[i] & (layer_2_pval_FDR < 0.05))
}
names(disease_significant_layer_2) <- disease_interest
disease_significant_layer_3 <- list()
for(i in 1:length(disease_interest)){
disease_significant_layer_3[[i]] <- logrank_pval_df %>% filter(disease == disease_interest[i] & (layer_3_pval_FDR < 0.05))}
names(disease_significant_layer_3) <- disease_interest
disease_significant <- list()
disease_significant[["layer_2"]] <- disease_significant_layer_2
disease_significant[["layer_3"]] <- disease_significant_layer_3
ここまでの解析を改めて実行しようとしても、かなりの時間が必要になる。もはやデータをロードするだけでもオーバーナイトする勢いである。なので、重要なデータセットはRのデータセット(.rds)や、TSVファイルで保存しておく方が良さそうである。これで、必要に応じてこれらを読み込むことで、全部のデータをロードするより早く、必要なデータを読んでくることが出来る。
# code 70
saveRDS(logrank, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/logrank.rds")
saveRDS(logrank_pval, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/logrank_pval.rds")
write_tsv(logrank_pval_df, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/logrank_pval_df.tsv")
saveRDS(disease_significant, "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/disease_significant.rds")解析環境を記録しておく
もし論文や報告書を作成する場合は、これもどうしても必要になる。それに、最後に実行した日はいつだったかを記録しておくのも、自分の防備録として非常に有用である。なので最後に実行した日とsessionInfo()をファイルに出力しておく。
# code 71
file.remove("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/save_log.txt")
file.remove("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/sessionInfo.txt")
cat(paste0("The analysis was saved at ", Sys.time(), "\n"),
file = "/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/save_log.txt")
sink("/mnt/team4tb/Dropbox/Blog/2025 10 24 TCGA analysis/sessionInfo.txt")
sessionInfo()
sink()まとめ
以上が、自分が行ったTCGAのデータを使って、ある遺伝子の発現量やそれらが関係する遺伝子セットが患者の生存率に影響するかどうかを解析する方法である。
この解析の注意点としては、やはりカプランマイヤー法による生存率解析(生存期間解析)で使った観察期間である。今回は、TCGAに登録された(そして公的にアクセス出来る)症例の全観察期間で打ち切りを設定したが、ここは5年で区切って打ち切りを設定した方がよりリーズナブルかもしれない。ここはcode 54の最後にコメントアウトしてあるので、必要ならばこれを使うべきである。また、正常組織の例数の少なさにも注意する必要が有る。やはり、腫瘍組織の症例数は130例とかあるののに、正常組織の例数は10例とかだと、検出力が足りないようにも思う。そういう意味では無理して正常組織は含める必要はなかったかもしれない。
この後の解析、というか、作業としてはShinyでいつでも解析結果にアクセス出来るようにすることと思う。これは今後やって行こうと思う。これにより、急に必要になった場合はShinyで表示すれば、誰かと共有できるはず。ここまでできれば、このデータも重宝するかと思う。
ここからのRのダウンロード(文章を読みたくない人用)3
2026/01/12(月)追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う
上記で、RでAnndataを使おうとすると結局Reticulateパッケージを使う羽目になってしまい、それだったらRでやるわ、という話を書いたが、そう言えば、Rでもここで取り扱っている遺伝子発現量に症例毎の診療情報やシークエンスに使用されたサンプルの情報を効率良く扱うことが出来るパッケージがあった。SummerizedExperimentである。これを使えば、Anndataのようなデータの構造で解析を行うことが出来ることに気がついた。SummerizedExperimentは、まさにAnndataのように行と列の順番が一致している必要が有るが、それをうまいことリンクさせてそのような順番に依らないようにしたフォーマットのMultipleAssayExperimentというパッケージもあるらしい。これらを上手に使うことで、メモリを節約しながら解析を進めることが出来るはず、と思っている。