2025/12/14(日);初稿
2025/12/29(月);訂正;カプランマイヤー法による生存率の解析;教科書(生存期間解析、ISBN978-4-254-12861-1)に従って、打ち切りの設定を確認し、生存曲線(カプランマイヤー推定値)の横軸を1825日(5年)に限定した。
2026/01/03(土);訂正;元の記事のタイトル「TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.41, 2024)(正常組織のバイオリンプロットなし、GDC portalからのデータのダウンロード方法なし )」が長すぎてプラグイン「codoc」が正常に動作しなくなる可能性があったので、現在のタイトルに変更した。
2026/01/12(月);追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う。
- 1 はじめに
- 2 解析の流れ
- 3 注意点1
- 4 注意点2
- 5 注意点3
- 6 使用するパッケージ
- 7 RNA-seqのカウントデータを読み込む
- 8 ここまでのRのダウンロード(文章を読みたくない人用)1
- 9 サンプル情報と診断情報を読み込む
- 10 カウントデータ、メタデータ、診断情報を関連付ける
- 11 データをAnndataで保存する(Optinal)
- 12 ここまでのRのダウンロード(文章を読みたくない人用)2
- 13 解析のためのデータを読み込む
- 14 Geneset enrichment analysis (GSEA)に使用するGMTファイルを作成する
- 15 Geneset enrichment analysis (GSEA)に使用するGCTファイルを作成する
- 16 症例毎にGSEAを行ってエンリッチメントスコアを計算する
- 17 各種GSEAの結果をまとめる
- 18 疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する
- 19 疾患毎の遺伝子発現量やエンリッチメントスコアをバイオリンプロットで比較する
- 20 カプランマイヤー法による生存率の解析
- 21 データセット全体を保存する
- 22 ここまでのRのダウンロード(文章を読みたくない人用)3
- 23 追加の解析
- 24 まとめ
- 25 2026/01/12(月)追記;AnndataではなくSummerizeExperimentやMultipleAssayExperimentが使えると思う
はじめに
以前、TCGA(The Cancer Genome Atlas)に登録されたデータをダウンロードしたが、ここではTCGAのデータを使って各疾患毎の患者の生存率をカプランマイヤー法により解析する方法を述べる。これらはがんや腫瘍の遺伝子発現や遺伝子変異とそれに紐づく患者の情報であり、他の解析(基礎実験など)で疾患に関連しそうな遺伝子やタンパク質が見つかった場合、それらと疾患との関連性、特にそれらが患者の生存率に影響が及ぶかどうかを解析することが出来る。
解析の流れ
TCGAなどの解析で非常に面倒なのか、RNA-seqのカウント値が記載されたtsvファイル(のファイル名)と、それに対応する個々の患者の情報を結びつける作業が必要なことである。そのために、列名を適切に直し、不必要な重複を取り除かなければならない。このデータのクリーニングとも言える作業の後に、カウント値を種々の手法を使って解析し、最後に全解析結果と患者の情報を結合する。ここでは、そのカウント値を使ってGSEAなどを行い、遺伝子発現プロファイルの特徴を各患者(症例)毎に求め、それらを集計、必要ならば統計解析し、最後にそれらの特徴からある遺伝子や遺伝子セットに着目した生存率を求めて、関心のある遺伝子の発現量や遺伝子セットのエンリッチメントの程度が、患者の生存率にどれだけ関連するかを解析する。
注意点1
同じような記事(後日にポストした記事・新しい記事)を別に書いている。この記事との内容の違いは以下の表の通りである。もしこの記事を購入する場合は、以下の表を確認して、どちらが必要なのかを判断していただきたい。比較対象は新しい記事であるが、その解析に使ったTCGAのデータのバージョン(v.43)は、この記事のバージョン(v.41)よりも新しい。ただし、症例数はそんなに大きく変わらなかった。新しい記事にはssGSEA2とfgseaによるエンリッチメント解析が載っていない。この記事ではMsigDBのH: hallmark gene setsだけなのに対し、新しい記事ではMsigDBに登録のあるヒトの遺伝子セットを全部使ってエンリッチメント解析を行っている。また、新しい記事では正常組織のバイオリンプロットとの比較があったり、患者の生存期間に統計的有意な影響を与える遺伝子をまとめていたりするので、参考にはなると思う。
| 項目 | この記事 | 別の記事(後日にポスト) |
|---|---|---|
| タイトル | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.41, 2024) | TCGAのデータとRを使ってがんや腫瘍の遺伝子発現量と患者の生存率の関係を解析する(TCGA, v.43, 2025) |
| TCGAのバージョン | v.41, 2024 | v.43, 2025 |
| GDC portalからのデータのダウンロード方法 | なし | あり |
| GMTファイルの作り方 | あり(ただし、Geneontologyから選んできた遺伝子セット1つだけ) | なし |
| 使ったGMTファイル | Geneontologyから選んできて自分で作成したHRR(Homologous Recombination Repair)と、MsigDBのH: hallmark gene sets(Human MSigDB v2025.1.Hs、2025年10月時点)のみ。 | MsigDBに登録されているヒトの遺伝子セット全部(Human MSigDB v2025.1.Hs、2025年10月時点) |
| エンリッチメント解析 | ssGSEA2 escape GVSA fgsea | GSVA escape |
| バイオリンプロットによる疾患毎の遺伝子発現量もしくは遺伝子セットのエンリッチメントの比較 | 常組織なし ウィルコクソンの順位和検定なし | 正常組織あり ウィルコクソンの順位和検定あり(ただし、正常組織の例数が腫瘍組織に比べて少ないのでちょっと注意が必要と思う。) |
| 生存期間解析でのログランク検定 | あり | あり |
| その他 | 比較するデータの数(リストの要素の数)がそんなに多くないので、データの準備がそんなに大変じゃない。 | 比較するデータの数(リストの要素の数)が多く、そのために少しデータの準備がすこし複雑でメモリを喰いまくる。 |
注意点2
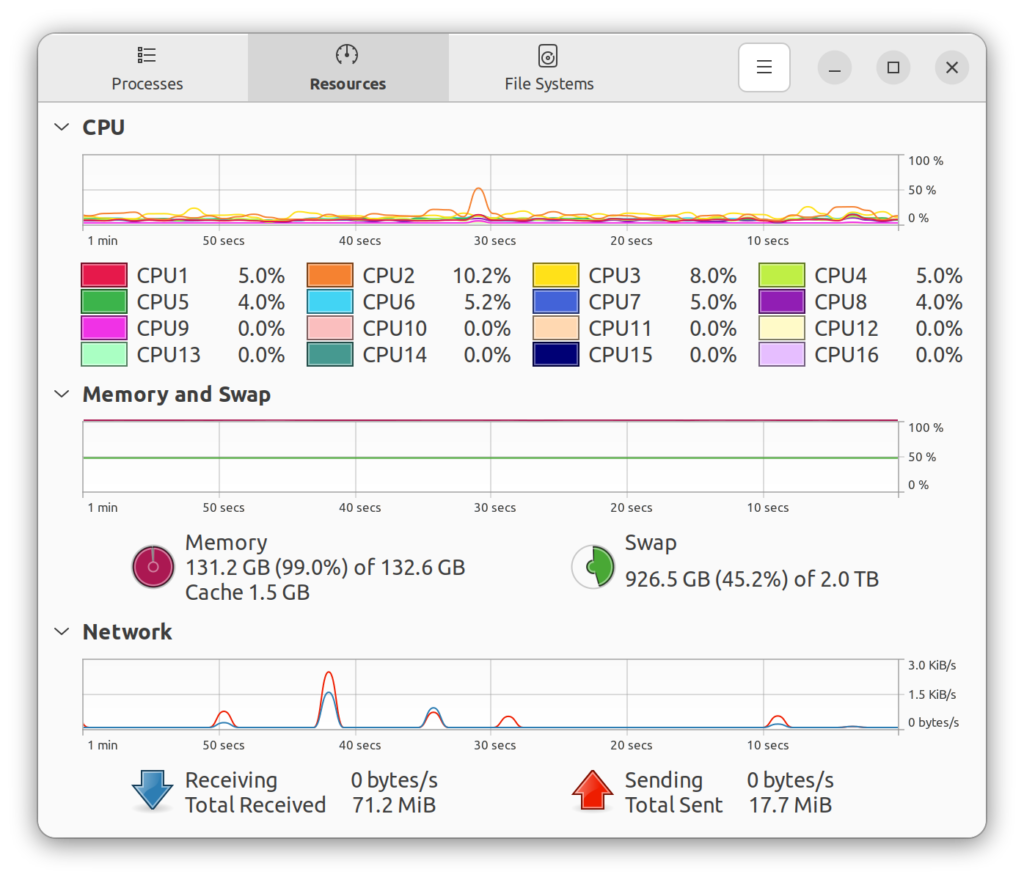
この解析では要求されるコンピューターの性能は案外高い。この解析で使用したCPU、RAM(Random Access Memory)(物理メモリ)のサイズ、SWAPのサイズは、それぞれRyzen 9 9950X、128GB 、2TBである。特に、RAMのサイズが大分と効く。もはや256GBあっても十分ではない。なので、SWAPを充分に用意しておかなくてはならない。SWAPについては、以前cellrangerを使っているときにSWAPファイルだとどうしてもメモリリークみたいなことになっていたので、2TBのm.2全部をSWAPパーティションとして与えている。こんな無駄に大きい理由は、本来別の用途でこの2TDのm.2を購入したのだが、結局本来の用途に不要になってしまい、それを流用したためであり、大きいからと行って無駄に1TBにSWAPパーティション、1TBにデータの保存とか設定するのが面倒だったからである。言うても、一般的には必要ではないのだが、逆にこれだけあればメモリについては不安になることもない(SWAP Thrashing;スワップスラッシング問題はある)。これが一杯になる計算があったとしたら、その計算では何かしらのエラー(例えば、スパースマトリックスとして扱うべきものを、普通のマトリックスとして計算してしまったとか、一つあたり100GB以上もあるデータセットに対する条件分岐をforeachで50000回も回してしまったなど。これについては何かしらの方法がありそうだけど、自分はそのあたり良くわからん。)が出ている可能性があるし、エラーではないとしたら自分が知っているものでは、シングルセルRNAシークエンスの解析においてtradeseq(あるクラスター間のトラジェクトリーに沿った遺伝子発現解析に使用するパッケージ)くらいである。データセットのサイズが大きいので、RAMの容量は多い方が良い、というか、少なすぎるとTCGAのような大きいデータセットの取り扱いは無謀と言える。以前も書いたが、これはRの弱点と思う。Rは計算しようとするデータセットを全部RAMに入れて計算しようとするので、例えば、Rではデータセットのサイズが500GBならば、RAMはシンプルに500GB以上必要になる。こういう場合、コンピューターに積んであるRAMの容量が16GBとかで、SWAP(Windowsでは「仮想メモリ」と呼ばれている)を500GBとか確保しないと、データがRAMに入り切らずに、入ったとしても計算出来ずにRもしくはコンピューターがクラッシュすると思う。また、こんなに大きなデータセットに対してGSEA(Gene Set Enrichment Analysis)等を行う場合は、ある程度性能の高いCPUも必要である。これがなければ、計算は出来るがそれに1ヶ月費やす、とかになる。
また、自分はこれらの解析をなんとCore i5、16GBのSurface Pro 8(5年以上前のコンピューター)でやっていたが、この手のCPUファンなどの冷却機能が劣るコンピューターを使うときは要注意である。Surfaceなどはファンはあるにしても、コンピューターの背面全体も使って熱を放出するような、なんとも受け身な冷却方法を採っており、これだと驚くほどコンピューターが熱くなる。こういったラップトップでここに書くような解析をしてしまうと、熱でラップトップの寿命を縮めるのは間違いない。
個人的にはメモリと冷却が十分であれば、5年以上前のコンピューターでも十分に解析に使用出来ると思っている。むしろ、CPU等は多少古いほうがパッケージとの相性などは良い可能性もある。もちろん、少し古いコンピューターではRAMが詰めなかったりするので、その場合はサーバーとかになるのだろう。所属する研究室や会社が許せば東大のSHIROKANEとか使ったほうがコストパフォーマンスが良いような気がする。その場合は自分で構築していいのか確認しなければならないような気がする。
また、ハードウェアで最近思うこととして、Rだけを使うならばIntelのCPUでWindowsを使うのが一番良いのではないかと思う。最近ではインテルへ不信感とCPUの性能の悪さからAMDが使用されがちであるが、これらのパッケージが開発・作成されたときに用いられていたCPUはインテルであり、インテルのCPUの方が相性が良いように思う。最近のMacは使ったことがないのでよくわからない。一方、自分はダウンロードやインストールが楽なのでUbuntuを使う。結局、なんやかやで痒いところに一番手が届くのがUbuntuである。
注意点3
この解析は、「疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する」の章までは本当に面白くない。それまではデータを読み込んだり、クリーニングしたり、整えて結合したりするだけである。途中のssGSEA2、escape、GSVA、fgseaを使ったGSEA(Gene Set Enrichment Analysis)があるが、整えたマトリックスなりGCTファイルを関数に入れるだけである。実際に解析やプレゼンテーションに使えそうな結果は、「疾患毎の遺伝子発現量とエンリッチメントスコアをヒストグラムで表示する」以降からである。
以下、順次解析を始めていく。ちなみに、以下はすべてUbuntu上でのコードであるが、おそらくMacでもほぼ同じコードが動くはずである。Windowsの場合はパスの書き方が違うので注意が必要である。おそらく正規表現をエスケープさせる必要がある。なお、カプランマイヤー法云々はここでは述べないので、それらを知りたい場合は他のウェブページ、もしくはChatGPTにでも聞いたほうが色々と知ることが出来ると思う。
途中、Rのコードをダウンロード出来るようにしたので、読むのが面倒な場合はそのコードをダウンロードして、ディレクトリを適宜変えながら解析すれば良いと思う。また、インストールするパッケージ、使用しているコンピュータ、OSによってはエラーが出るときもあることを了承いただきたい。連絡をくれれば「出来ることであれば」サポートしようと思っている。
使用するパッケージ
以下のパッケージを読んでおけば、遺伝子発現量の解析であれば十分である。自分はこの環境で遺伝子発現の解析を行なっている。ここに、適宜必要なパッケージを追加しながら、解析を進めていく。
基本はtidyverseでデータを整えて、survivalでカプランマイヤー法を計算し、survminerで可視化する、という流れである。可視化にはgridExtraで各グラフをパネルにする。またdata.tableは作成したデータの読み込みに使用する。生存率の解析としては、基本的にこれくらいしか使わない。これらに加えて、fread、doParallelなどは読んでおいた方が良い。これらはfgseaのように、通常ならばシングルコアで計算する(実はBiocParalelが使用されている。全く調整できない、もしくは、自分にそんなスキルはないので、非常に邪魔くさい。)けど、だからこそ並列化した場合には効果が絶大と言える。もしシングルコアで計算しようものなら、一つのコードチャンクに1週間とか費やす羽目になってしまう。他のパッケージで重要な物は、GSEAやエンリッチメント解析に使用する物、ヒートマップ作成に使用する物などである。
また、この解析では最後にSys.getenv()で、Rの環境変数を出力している。理由はこの解析では途中にダウンロードして整形したTCGAの各患者の遺伝子発現プロファイルに対しssGSEA2、escape、fgsea、GSVAを適用するが、そのときに並列化がうまく行かず、そのトラブルシューティングで環境変数を確認したためだ。通常ならばこれは不要だが、別に最初に表示しない理由もないので今回は確認した。
# DEG
library(baySeq)
library(edgeR)
library(DESeq2)
library(limma)
# enrichment analysis
library(clusterProfiler)
library(fgsea)
library(ssGSEA2) # devtools::install_github("nicolerg/ssGSEA2") # devtools::install_github(broadinstitute/ssGSEA2.0) and devtools::install_github(broadinstitute/ssGSEA2) did not work.
library(escape)
library(GSVA)
library(pathview)
# immune cell deconvolution
library(ConsensusTME)
library(estimate)
library(immunedeconv)
library(MCPcounter)
library(msigdbr)
library(quantiseqr)
library(TIMER) #devtools::install_github('hanfeisun/TIMER'); it will not work!! use TIMER of immunodeconv if you need.
library(xCell)
# stats
library(NSM3) # sudo apt install libgmp-dev libgmp10 libgmp3-dev
library(pwr)
library(missForest)
# basic
library(beeswarm)
library(circlize)
library(org.Hs.eg.db)
library(org.Mm.eg.db)
library(doParallel)
library(foreach)
library(parallel) # Need it for use of baySeq
library(pheatmap)
library(rtracklayer)
library(rvest) # this is for extraction of html table; results of leading edge analysis.
library(xml2) # this is for extraction of html table; results of leading edge analysis.
library(openxlsx) # for read xlsx file.
library(ComplexHeatmap)
library(tidyverse)
library(data.table)
library(survminer)
library(survival)
library(gridExtra)
library(MASS)
library(coin)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(RhpcBLASctl)
# # shiny
# library(shiny)
# library(bslib)
# Check global environment
Sys.getenv()