2026/05/11(月);初稿
2026/06/13(土);記事が長すぎてCodecプラグインが動かなかったので、コードをSTORESで販売することにした。
- 1 はじめに
- 2 注意点1.RNA-seqとタンパク質(ペプチド)質量分析のデータの違い
- 3 注意点2.解析に使用できるタンパク質の数、疾患の種類、症例数がTCGAよりも少ない
- 4 注意点3.疾患ごとのタンパク質発現量の比較と生存率の解析方法はTCGAとほぼ同じ
- 5 使用するパッケージ
- 6 ここから使用するRのダウンロード1
- 7 使用するデータ
- 8 使用するデータを読み込んで整える
- 9 CPTACのタンパク質発現量は中央値で正規化されている
- 10 missForestによる欠損値の補完(imputation)
- 11 タンパク質発現プロファイルと臨床情報を結びつける
- 12 escape、gsva、fgseaを使ってGSEA(Gene Set Enrichment Analysis)を行う
- 13 疾患名の整理
- 14 タンパク質発現量やエンリッチメントスコアのヒストグラムを表示してみる
- 15 バイオリンプロットで疾患ごとのタンパク質発現量を比較する
- 16 生存解析
- 17 タンパク質発現量と遺伝子発現量の相関を解析する
- 18 よくわからないタンパク質発現プロファイルの約90%が韓国と中国の研究機関発だった
- 19 まとめ
はじめに
これまでTCGA(The Cancer Genome Atlas)に登録された各種がんの遺伝子発現プロファイルと、その患者の生存率(生存期間)を解析してきた。この記事ではCPTAC(Clinical Proteomic Tumor Analysis Consortium)に登録されたデータを使ってがんや腫瘍のタンパク質発現プロファイルとその疾患の臨床情報を用いて、特定のタンパク質の発現量と患者の生存期間の解析をカプランマイヤー法で解析してみようと思う。
このCPTACは、種々のがんのタンパク質発現プロファイルを質量分析(Mass Spectroscopy;MS)を使って取得し、その患者に関わる臨床情報と共に登録した公開データベースである。言ってみればTCGAのタンパク質バージョンである(CPTACでプロジェクトは別物、解析自体も別物だけど)。未だ発展途上であり、登録されている疾患の数や症例数はTCGAほど多くないし、MSのデータのクオリティーも遺伝子発現ほど良くないだろうし、CPTAC側としてもデータのインテグリティーに問題があり、特にタンパク質発現プロファイルとそのファイルとリンクする臨床情報を結びつけることができない症例が有ったりする(正直これはCPTACではなく、こういうことをやってデータを無駄にするような大学や研究機関が間抜けで無責任なのだが。これは解析のときに述べる)が、このCPTACにはプロテオームだけでなく、ホスホプロテオーム、メタボローム、アセチローム、ユビキチノーム等のデータが登録されており、そんな欠点を補って余りある有用なデータベースである。昨今、そしてこれからのトランスレーショナル研究には必要不可欠なのではないかと思っている。また、同じ症例の遺伝子発現プロファイルがTCGAに登録されていたりするので、遺伝子発現とタンパク質発現の相関なんかも解析することができる。これを直ぐに使える形で解析したデータを持っておけば、とても便利だろうと思っている。
2026年3月になってようやくサーバーが正常に動くようになっていたので、後日、新しい版のデータでも別に解析してみようと思う。言うて、解析手法はここやこことほとんど同じはずであり、手法としてはそこまで難しくない。それに、データが少し古いからと言っても、症例数はそこまで急に増えるようなことはないので、この解析方法や解析したデータは結構長く使えるのではないかと思っている。
余談だが、生存率解析ではなく生存期間解析や単に生存解析の方が正しい呼称だと思う。言い方としては、例えば「生存解析(survival analysis)を行った結果、overall survivalの中央値は6ヶ月です。」とかになるし、overall survalの日本語約は「全生存期間」だったりするためだ。ただし、誰にでも通じる言い方として生存率解析と言ってしまうことも多いと思う。この記事では、タイトルにこそ生存率の解析としているが、本文中では生存期間解析や生存解析と言うことにする。
注意点1.RNA-seqとタンパク質(ペプチド)質量分析のデータの違い
個人的に重要と思うことが、RNA-seqのデータとMSのデータの違いである。これまではRNA-seqの技術が圧倒的に早くに発展、普及してきたので、この手の解析は遺伝子発現プロファイルに基づくことが多かった。そして、その勢いでMSのデータを解析すると、おそらくどこかで躓くような気がする。RNA-seqの解析から入った人間は、MSのデータの性質がRNA-seqと大きく異なる事を忘れてはならない。RNA-seqの場合、昨今ではハイスループットシークエンサーが利用されている。これは読んできた(リードした)シークエンスを、ゲノム(というかエクソン上)にマップして、ゲノム上にどのくらいマップされたのかカウントしていく。データは「カウント値」として記録されるので、二項分布(負の二項分布というヤツ)に従う。それに、ゲノム上にどのくらいマップされなければならないかをカバレッジなどの指標にしてシークエンスを計画するし、ゲノム上に似たような配列があれば、そこにマップされてしまったりと、けっこうな領域をカバーすることができる。それにデータがカウント値であることから、欠損値(欠損値というのかはわからん)が出た場合は、それをカウント値がゼロとして解析を行うことができる。一方、MSのデータは、質量分析の結果でてきたフラグメントのシグナル強度を計測し、それをデータベースに照合して解析する。これはカウント値ではない(非常に申し訳ないが、自分はこのくらいしか知識がない。chatGPTの方が詳しく教えてくれる)ので欠損しているタンパク質をカウント値がゼロと見なすことが難しいし、検出されてこない原因が単に発現が低いのか、技術的に拾ってくるのが難しかったためなのか、本当に発現していないのかよくわからない。また、欠損値をカウント値がゼロとみなして解析するにしても、欠損値が多すぎるような気もする。統計分布と欠損値の取り扱いが異なるので、RNA-seqの解析でよく利用されるDESeq2やedgeRはDEG(Differential Expressed Gene)の解析には不向き(使えないこともないが。)であり、他の適切なパッケージを利用するほうが良い。また、利用するパッケージにも依るが、解析するためには欠損値は他の値(欠損値ではない値)を参考にして補完(imputation)しなければならない。それに欠損値を補完するか自体よく考えた方が良い場合もあるように思う。言うて解析する側は、その目的、例えば、何らかの群同士を比較してDEGを出すのか、この記事での解析のように単に発現で分類して他の解析との関連をみるのかによって、適切な方法で解析を進めなくてはならない。
因みに、limmaとかなら使えると思う。limmqはもともとcDNAマイクロアレイのDEG用だったはずであり、これはcDNAマイクロアレイは蛍光シグナルを検出しているためだ。
後述するが、この記事ではDEGの検定などは行っていないので、そのためのそのようなパッケージは特に使用していない。しかし、欠損値の推定はゼロを使うよりは良いということで、missForestというRのパッケージを使って行っている。ここがRNA-seqを元にした解析と異なる点である。
注意点2.解析に使用できるタンパク質の数、疾患の種類、症例数がTCGAよりも少ない
はじめにで書いたように、CPTACは未だ展途上である。バージョンだってまだ5くらいであり、疾患の数がTCGAと比較しても少ない。特に痛いと思うのがこの点で、この解析でも最終的に11疾患くらいしかなかった。依って、解析に必要な疾患のデータが十分でなかったり、そもそも関心のある疾患が無い可能性もある。その場合は他のCPTACだけでなく他のプロジェクトも使用する必要が出てくるが、プロジェクトを横断する場合は登録するデータの違いなども問題として出てくるに違いない(知らんけど)。それに加えて、タンパク質発現プロファイルのデータと、患者の情報をうまく結合できない症例もある。このあたりを考えて、CPTACやPDC(Proteomics Data Portal)に登録のあるデータを使用する必要がある。
注意点3.疾患ごとのタンパク質発現量の比較と生存率の解析方法はTCGAとほぼ同じ
タンパク質発現量と患者データの確認や結合の方法は、詳細は違えど結局似たようなことをやるし、疾患ごとのタンパク質発現量の比較や生存率の解析方法は以前にTCGAのデータを解析したときとほぼ同じである。
あと、この記事データのクリーニングとか整形ばっかりで本当に面白くない。9割がデータのクリーニングである。もし買う人がいたら、本当に注意したほうが良い。本当に面白くない。むしろ苦痛である。
早速以下からデータの解析を始める。
使用するパッケージ
以下がこの解析で使用するパッケージである。上記で「RNA-seqとデータ取得の方法や考え方が違うから注意」とかなんとか言っておきながら、いつもRNA-seqの解析で使用している基本パッケージを使用した。以下のコードにDEGとか初っ端に書いてあるけど、ここではDEGは行わないのでこれらは使用しない。一方、エンリッチメント解析のパッケージであるfgsea、escape、GSVA、並列化に関するパッケージforeach、doParallel、欠損値imputationにmissForest、生存率解析用にsurvminer、データの読み込みにdata.table、tidyverse、gridExtra等を使用する。以下を読み込んでおけばこの記事の解析は一通りできるはずである。ここで使用するエンリッチメント解析のパッケージであるescape、gsva、fgsea、そしてここでは解析に費やす時間の関係上実際には使用しないが(動くコードは書いた)、ssGSEA2は、本来遺伝子発現解析のために使用されると思うが、エンリッチメント解析としてやっていることはタンパク質発現解析にも利用出来るはずである。実際、ssGSEA2はPTM(Post Transcriptional Modification)の解析にも使用されているようである(やったことはないが)。
# DEG
library(baySeq)
library(edgeR)
library(DESeq2)
library(limma)
# enrichment analysis
library(clusterProfiler)
library(fgsea)
library(ssGSEA2) # devtools::install_github("nicolerg/ssGSEA2") # devtools::install_github(broadinstitute/ssGSEA2.0) and devtools::install_github(broadinstitute/ssGSEA2) did not work.
library(escape)
library(GSVA)
library(pathview)
# immune cell deconvolution
library(ConsensusTME)
library(estimate)
library(immunedeconv)
library(MCPcounter)
library(msigdbr)
library(quantiseqr)
library(TIMER) #devtools::install_github('hanfeisun/TIMER'); it will not work!! use TIMER of immunodeconv if you need.
library(xCell)
# stats
library(NSM3) # sudo apt install libgmp-dev libgmp10 libgmp3-dev
library(pwr)
library(missForest)
# basic
library(beeswarm)
library(circlize)
library(org.Hs.eg.db)
library(org.Mm.eg.db)
library(doParallel)
library(foreach)
library(parallel) # Need it for use of baySeq
library(pheatmap)
library(rtracklayer)
library(rvest) # this is for extraction of html table; results of leading edge analysis.
library(xml2) # this is for extraction of html table; results of leading edge analysis.
library(openxlsx) # for read xlsx file.
library(ComplexHeatmap)
library(tidyverse)
library(data.table)
library(survminer)
library(survival)
library(gridExtra)
library(MASS)
library(coin)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(RhpcBLASctl)
library(psych) # for geometric mean
# # shiny
# library(shiny)
# library(bslib)
# Check global environment
Sys.getenv()ここから使用するRのダウンロード1
以降のコードは、以下のリンクで販売している。ZIPを展開するとRのコードがいくつかあるので、そのうちの1 PDC expression matrix.Rmdというファイルが、以降のコードである。
文字を読みたくない人やRを読める人は、それをコードをダウンロードし、それを何らかのエディターなり開発環境で開いた方が良いと思う。
使用するデータ
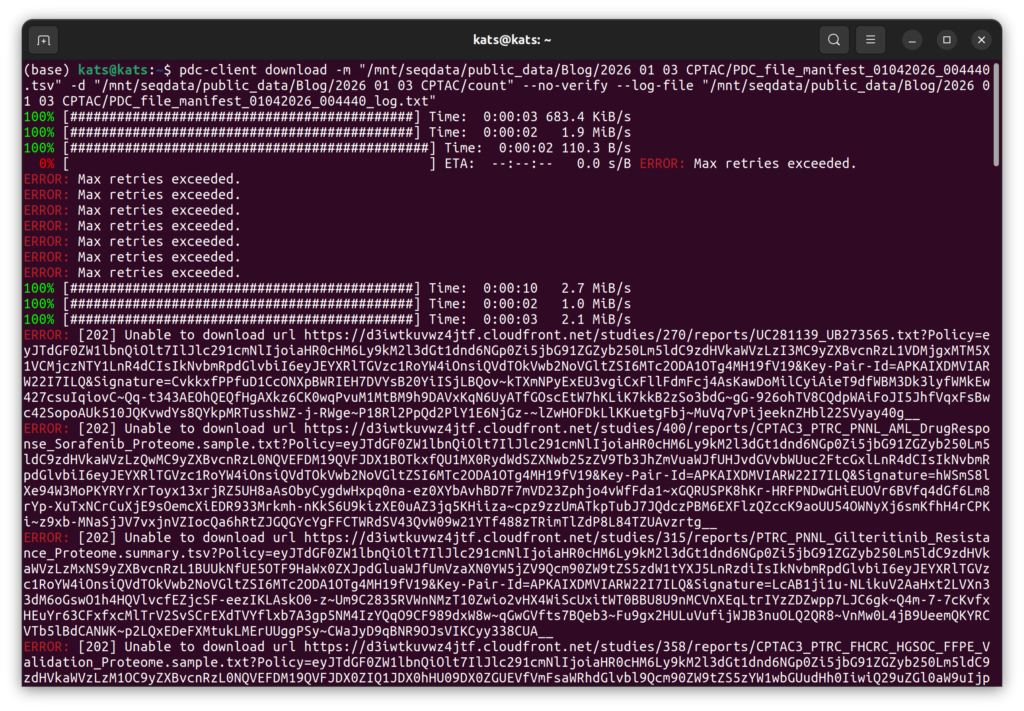
データとして以前の記事のためにダウンロードしたものを使用する。その理由は、最新のデータをダウンロードしようとしても、サーバーがうまく動いていないようだったためである。2026年1月の時点で最新のデータで解析をしようとしたのだが、以下の画像のようにエラーが出まくった。pdc-clientもちゃんとインストールできているようだったし、おそらくサーバーが不調だったと信じている。そして、上述の通り2026年3月になるとダウンロード出来始めたので、2026年3月時点のデータの解析は別の記事にしようと思っている。とりあえず、少し古くなるがv.4.4のデータで解析を進めることにする。注意点3.にも書いたが、この解析は列名が変わったとしてもデータの整理が大変になるくらいで、発現量の比較や生存率の解析自体はTCGAのそれと大きく変わらない。だから、ここで述べる解析も最新のデータに対応するはずである。実際、これは以前にTCGAの解析で使ったコードをそのまま流用しているが、特に大きな問題はなかった。そういうことで、このCPTACの解析方法をTCGAに用いたり、逆にしたり出来るはずである。解析として異なるのは、やはりデータの整理の部分であった。
使用するデータを読み込んで整える
code 1では、ダウンロードしたデータのファイルパスをlist.files()で読み込んできて、それらをリストCPTACの各要素に入れる。
# code 1
CPTAC <- list()
CPTAC <- list.files(path = "/mnt/seqdata/public_data/Blog/CPTAC/count", all.files = TRUE, full.names = TRUE, recursive = TRUE)code 2では、取得したパスをデータフレームCPTAC_dfに変換する。次に、各行のファイルパスから、tmtもしくはitraqで得られたペプチドのシグナル強度の部分を取ってくる。中にはリン酸化タンパク質のシグナル強度のデータも含んでいたりするので、それを一旦取得してから、改めてそれらを除外している。これらはstr_detect()に正規表現を入れて抽出してきている。正規表現は自分で考えるよりchatGPTに教えてもらったほうが正確に記述できる。なので、自分は正規表現を端から自分で考えるつもりはない。一応、最後にforeach()でファイルパスを出力してざっと眺めてみた。
# code 2
CPTAC_df <- unlist(CPTAC) %>% data.frame()
colnames(CPTAC_df) <- "path"
all_count_file <- CPTAC_df[str_detect(CPTAC_df$path, "tmt.*\\.tsv$")|str_detect(CPTAC_df$path, "itraq.tsv$"),] %>% data.frame()
colnames(all_count_file) <- "path"
proteome_and_peptide_count_file <- all_count_file[str_detect(all_count_file$path,"^(?!.*phospho).*"),] %>% data.frame()
colnames(proteome_and_peptide_count_file) <- "path"
proteome_count_file <- proteome_and_peptide_count_file[str_detect(proteome_and_peptide_count_file$path,"^(?!.*peptide).*"),] %>% data.frame()
colnames(proteome_count_file) <- "path"
# just check the file extention of iTRAQ data.
# itraq <- IRAKM_CPTAC_df[str_detect(IRAKM_CPTAC_df$path, "itraq.tsv$"),]
foreach(i = 1:nrow(proteome_count_file)) %do% {print(proteome_count_file[i,])}TCGAでは、1つ症例の遺伝子発現プロファイルは1つのファイルで保存されていた。一方、CPTACでは、1つのプロジェクトで得られた複数の症例のタンパク質発現プロファイルが、1つのTSVファイルにまとめられている。なので、1つのプロジェクトのTSVファイルを読むと、列が各症例、行が参照されてきたタンパク質で構成された複数症例のタンパク質発現プロファイルを得る事が出来る。
目的のファイルのパスが集まってきたら、それをforeach()で読み込んでいく。これに%dopar%を入れると、何らかのエラー(少ない数だとちゃんと動くので、メモリのオーバーフローとか、そういうヤツだと思う)で止まってしまう。なので、%do%を使っている。自分は詳しくわからないのだが、foreach() %do% {}は、for() {}より少し早いようだ。おそらくCPUの使われ方が違うのだろうと思う。しかしながら、foreach()はdoParallelで並列化を管理しないと、どこかでクラッシュするように思う。なので、安全を期すなら、for(){}にした方が絶対に良い。かなり遅いが。
読み込んできたファイルはリストproteome_collectionの各要素に入っており、このリストの各要素はプロジェクトごとに別れている。一つのプロジェクトには複数の症例からのタンパク質発現プロファイルが入っているが、このままではファイル数がパッと確認出ない。なので、一旦それらを全部結合し、ファイル数を確認する。このファイルを各々の症例に結びつけていくので、この数が処理をする前の一番最初の症例数にほぼ等しいことになる。リストproteome_collectionの各要素に入ったデータフレームをreduce()とinner_join()を使って結合していく。reduce()に渡すinner_join()は、パラメーターを設定できないので、予め列Geneで結合出来るように新しく関数inner_join_for_check_file_numberを設定し、それを使って結合し、その列数を取得する。
# code 3
# Important
# Need to connect E drive.
# Read the dataset of PDC mass spec data for all patients, which are associated with "IRAK3".
proteome_collection <- list()
foreach(i = 1:nrow(proteome_count_file), .packages = 'readr', .combine = 'list') %do% {
proteome_collection[[i]] <- read_tsv(proteome_count_file[i,])
}
# Need to check how many files are stored in proteome_collection.
# The data frame "file_number" is for just checking file number. It is not recqured for other analysis.
view(proteome_collection[[1]])
inner_join_for_check_file_number <- function(x, y) {inner_join(x, y, by = "Gene")}
file_number <- purrr::reduce(proteome_collection, inner_join_for_check_file_number)
ncol(file_number)code 4では、リストproteome_collectionに入れた各データフレームから、不要な列を削除していく。NCBIGeneID、Authority、Description、Organism、Chromosome、Locusは不要である。ここでもちょっとでも早く終わるよう願いを込めてforeach()を使う。そして、その中でselect()を使うので、foreach()内で使用するパッケージとしてdplyr(とpurrr)を設定しておく。これで、不要な列をselect()で除外出来る。
注意点2.で述べたように、CPTACのデータは発展途上なので、一つのタンパク質発現プロファイルのに対して、複数の症例もしくはサンプル(正常と腫瘍のプールという全く意味のわからないspeciemen(解析に使用された試料もしくはサンプルのこと)を使っているプロジェクトがある)がある。後々、それらを除外するためには、PDCから始まりその後ろに番号が何桁か続くプロジェクト名が非常に有用である。データフレームproteome_count_fileの列pathから、「PDCから始まり、その次になんらかの数字が続く文字列」を、str_detect()を使って抽出してくる。そのためには正規表現で”PDC//d+(?=/)”を使用する。正規表現はchatGPTに聞いたら良い。それを、ファイル名の前に区切り文字をアンダーバーを使って付けておいた。こうすれば、ファイル名だけでどの疾患なのか区別出来ない場合、このプロジェクト名で判断できるようになる。余談だが、この解析ではどうしてもファイル名と疾患が一致している自信がない場合、そのファイルは最終的に解析から外した。そうしなければ、正確な解析が出来ないためである。 これをデータフレームtemp_dfとして、後ほど使用する。
# code 4
# Remove unnecessary columns from the dataset.
proteome_collection_wo_unnecessary_column <- list()
foreach(i = 1:(length(proteome_collection)), .packages = c('dplyr', 'purrr'), .combine = 'list') %do% {
proteome_collection_wo_unnecessary_column[[i]] <- proteome_collection[[i]] %>% dplyr::select(-one_of("NCBIGeneID", "Authority", "Description", "Organism", "Chromosome", "Locus"))
}
length(proteome_collection_wo_unnecessary_column) # 53
PDC_study_ID <- str_extract(proteome_count_file$path, "PDC\\d+(?=/)")
length(PDC_study_ID) # 53
names(proteome_collection_wo_unnecessary_column) <- PDC_study_ID
for(i in 1:length(proteome_collection_wo_unnecessary_column)) {
temp_df <- colnames(proteome_collection_wo_unnecessary_column[[i]])[2:ncol(proteome_collection_wo_unnecessary_column[[i]])] %>% data.frame()
colnames(temp_df) <- "file_name"
temp_df$PDC_study_ID <- PDC_study_ID[i]
temp_df <- temp_df %>% unite("file_name_W_PDC_study_ID", c("PDC_study_ID", "file_name"), sep = "_", remove = FALSE)
colnames(proteome_collection_wo_unnecessary_column[[i]])[2:ncol(proteome_collection_wo_unnecessary_column[[i]])] <- temp_df$file_name_W_PDC_study_ID}リストproteome_collectionの各要素のデータフレームを見ていると、その列名(ファイル名)に不要な文字が沢山入っていることがわかる。それを以下のcode 5で取り除く。おなしな文字をすべてアンダーバーに変換しておく。列名にスペースが入っていると、関数の中でそれを参照するときにいちいち” “や` `囲む必要があって使いにくいためである。多少見にくくなって判断に迷う場合もあるかもしれないが、コードを実行する上では問題は少ないように思う。
# code 5
# Clean up the data frame Column name contain a lot of unnecessary characters, and therefore replace them with "_".
foreach(i = 1:(length(proteome_collection)), .packages = c('dplyr', 'purrr'), .combine = 'list') %do% {
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("[- ]", "_", colnames(proteome_collection_wo_unnecessary_column[[i]]))
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("[/]", "_", colnames(proteome_collection_wo_unnecessary_column[[i]]))
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("[:]", "_", colnames(proteome_collection_wo_unnecessary_column[[i]]))
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("[()]", "_", colnames(proteome_collection_wo_unnecessary_column[[i]]))
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("[+]", "_", colnames(proteome_collection_wo_unnecessary_column[[i]]))
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("__+", "_", colnames(proteome_collection_wo_unnecessary_column[[i]]))
colnames(proteome_collection_wo_unnecessary_column[[i]]) <- gsub("[.]", "", colnames(proteome_collection_wo_unnecessary_column[[i]]))
}code 6で、出来上がったリストproteome_collection_wo_unnecessary_columnの各要素の入っているデータフレームを、結合する。結合にはfull_join()を使用する。reduce()にはfull_join()の引数を指定できないので、予め新しい関数full_join_by_geneを設定しておき、それを使用して結合する。それにより、データフレームmergedが出来上がる。
ここで、データフレームmergedの列名を「タンパク質発現プロファイル(ある症例から採取された試料やサンプルごとの全体的なタンパク質発現量のこと。遺伝子発現で言うところのライブラリのこと)」や「タンパク質発現プロファイル名」と呼ぶことにする。この先の解析でデータの整理で得られたデータフレームの列名に対しても「タンパク質発現プロファイル」や「タンパク質発現プロファイル名」を使用していく。
# code 6
# Merge All dataset by "Gene".
full_join_by_gene <- function(x, y) {full_join(x, y, by = "Gene")}
merged <- reduce(proteome_collection_wo_unnecessary_column, full_join_by_gene)
ncol(merged)
check_colnames <- colnames(merged)[2:length(merged)] %>% data.frame()
colnames(check_colnames) <- "name"CPTACのタンパク質発現量は中央値で正規化されている
CPTACのプロテオームのデータは、一番最初の3行が、各症例・試料ごとのタンパク質発現プロファイルの平均値、中央値、標準偏差である。それを別のデータフレームとして分けて保存する。このタンパク質発現プロファイルはCPTACに登録されるときにすでに中央値で正規化されているので、もしこれを正規化値に戻す場合は、medianの行にある値を使う必要がある。code 7では、これらの統計値が入っている最初の3行をデータフレームmerged_mean_median_sdとして、それ以外の値、すなわち正規化されたタンパク質発現プロファイルをデータフレームmerged_normalized_log2ratioとして分ける。そして、それらデータフレームの一列目の列Geneをそのデータフレームの列名とする。このとき、ペプチドをGene-centric(もしかしたらもとのタンパク質はスプライスバリアントとかだったかもしれないけど、それはもう区別せんでええ、同じタンパク質や、ということ。こういったフラグメントの解析では、本当のところはわからない)に集めてきているので、列名はGeneで良いか、と思ってGeneにしたと記憶している。今思ってみれば、紛らわしいからProteinとかにすれば良かった。
# code 7
# Extract statistics (mean, median and sd for each library calculated by PDC portal)
merged_mean_median_sd <- merged[1:3,]
# Remove statistics from the dataset.
merged_normalized_log2ratio <- merged[4:nrow(merged),]
# Add row name into statistics by PDC portal.
merged_mean_median_sd <- merged_mean_median_sd %>% column_to_rownames(var = "Gene")
# Add row name into the dataset.
merged_normalized_log2ratio <- merged_normalized_log2ratio %>% column_to_rownames(var = "Gene")code 8では、このデータフレームに入っているタンパク質発現プロファイルは一体どういった値なのかを確認する。全部の列を確認するわけにはいかないので、1列目を使って、この予め登録されている中央値がどのような計算によって求められているのかを逆計算してみる。
まず、データフレームmerged_normalized_log2ratioの1列目を取ってきて、ベクトルnormalized_log2ratioを作成する。CPTACによれば、どうやら中央値を使ってLog2正規化されているようなので、それを逆に計算していく。CPTAC側の計算は以下のようになっているはずである。
- normalized log2(ratio) = log2(each value/median) = log2(each value) – log2(median)
- log2(each value) = normalzed log2(ratio)+log2(median)
- 2^log2(each value) = each value
これをNA(欠損値)をそのまま除いて、each valueを求めてみる。そして、その中央値を求めてみる。それでCPTAC側が計算した中央値を求めることができれば、正規化前に戻せるはず。
ということでそれをやったのがベクトルbefore_normalized_log2ratio_wo_naである。その中央値を求めて見ると、どうやら-1.029058になる。おかしい…登録時の中央値-1.024735にならない…
次に、NAに0(ゼロ)を代入して同じ計算をしてみる。それがベクトルbefore_normalized_log2ratio_NA_is_0なのだが、この中央値を求めてみると-1.024735で、CPTACの計算と一致した。
ここで、疑問である。注意点2にイキって「欠損しているタンパク質をゼロとみなすことが統計的に難しい」という事を書いている。それは確かに正しいはず。でもCPTACは欠損値をゼロとして計算してやがる….
ゼロを入れても問題ないのはどんな場合だろうか….逆に、ゼロを入れてしまって問題な場合は、その発現プロファイルを統計的な分布にフィッティングして、そこから何らか値を推定するとききなはず。そしてそれ以外であれば、特にゼロを入れても問題がないような気がする….とは言え、一般的にはそれは問題な事がしられているので、ここではやっぱり何かしら科学的な方法で代入しなければならない。また、こうなってくるとなぜ何らかの値を代入するのか、という疑問にも至ると思う。代入しなくてはならない理由は、欠損値のままでは計算出来ない解析方法が沢山あるし、ある症例では欠損値なのに、他の80%の症例ではちゃんとした値が入っている場合など、どうしよう…となるからだろう。
# code 8
## Discrepancies in summary statistics at this url (https://pdc.cancer.gov/pdc-docs/data-analysis-guides#data-processing-and-harmonization) described CPTAC is using median normalization, and the values in data frame "merged_mean_median_sd" are a value before median normalization.
## The description is "The summary statistics (mean, median, standard deviation) in quantitation files are computed before median normalization. To match these values, add the median value back to each log2ratio column prior to performing statistical calculations."
## frequent question (https://proteomic.datacommons.cancer.gov/pdc/faq) discribed how to calculate back normalization. The discription is "Why don't the summary statistics (Mean, Median, StdDev) in the quantitation files match my calculations? The summary statistics in the quantitation files are computed before median normalization of the log2ratio values. To match these statistics, you need to add the median back to each column."
# these are the median and mean value by CPTAC
merged_mean_median_sd[2,1] # median = -1.024735
merged_mean_median_sd[1,1] # mean = -1.030376
# Each value without NA.
normalized_log2ratio <- merged_normalized_log2ratio[,1]
before_normalized_log2ratio <- normalized_log2ratio + merged_mean_median_sd[2,1]
before_normalized_log2ratio_wo_na <- before_normalized_log2ratio[!is.na(before_normalized_log2ratio)]
# Check distribution before substituting NA with 0
normalized_log2ratio_w_NA <- normalized_log2ratio
hist(normalized_log2ratio, breaks = 100, ylim = c(0, 14000))
merged_mean_median_sd[2,1] # -1.024735
median(before_normalized_log2ratio_wo_na) # -1.029058
mean(before_normalized_log2ratio_wo_na) # -1.039549
# NA replaced with 0
normalized_log2ratio[is.na(normalized_log2ratio)] <- 0
median(normalized_log2ratio) # 0
mean(normalized_log2ratio) # -0.005616025
before_normalized_log2ratio_NA_is_0 <- normalized_log2ratio + merged_mean_median_sd[2,1]
2^median(before_normalized_log2ratio_NA_is_0) # 0.4915004
# distribution if NA were sustituted with 0. The distribition was totally weird.
hist(normalized_log2ratio, breaks = 100, ylim = c(0, 14000))
merged_mean_median_sd[2,1] # -1.024735
median(before_normalized_log2ratio_NA_is_0) # -1.024735 # It is consistent with CPTAC calculation.
mean(before_normalized_log2ratio_NA_is_0) # -1.030351
# exp(mean(log(before_normalized_log2ratio_NA_is_0))) # NaN
geometric.mean(before_normalized_log2ratio_NA_is_0) # 0.2752213
## When 0 was substituted into NA in median normalized intensity by CPTAC or submitters, the median value was same as CPTAC's median. Following order of calculation was able to reproduce the median value by CPTAC or submitters.
# 1. Replace NA with 0 (Because the value should be centered with median value.)
# 2. Plus median (turned the value before normalization.)
# 3. Then median will be same as median by CPTAC. The result is log2ratio before normalization.
# 4. calculate power of 2. The result is ratio before normalization.
# 5. the value (intensity) before normalization
## If 0 was substituted into NA in median-normalized intensity, the data set provided by CPTAC, it means that the value was centered with median value.
# So, What meaning of NA in original dataset is very important. Why is NA generated? In mass spec data, NA means "Not detected" or "the value is under detection limit". The real value is unknown. In addition, each column has different median. what should be.code 9は、どのような値を使っていこうかと考えていたときに出したヒストグラムである。これは本解析には直接関係はない。データフレームmerged_mean_median_sdから、列名にUnshared_とある列以外を選び出し、そのうち中央値のみをベクトルmerged_mean_median_sd_shared_vectorとして作成し、そのヒストグラムを出してみた、というところである。その統計値がどんな範囲にあるのかをみるためにquantile()を計算している。
このUnshared_と記載のある値は、スプライスバリアントか何かで「固有の配列を持つペプチドとして検出された」というペプチド(タンパク質)である。これを解析に含めると、MSigDB
などで使用されているような一般的な遺伝子を使った分類ではなくなってくる(Gene-centricではなくなってくる)ので、そのような固有の配列を持ちそうなペプチドではなく、共通の部位として同定できたペプチド(タンパク質)であるsharedの記載がある列を使用して解析を行っていく。この解析ではその様に定める。
# code 9
# Following is stats of median value used for normalization at CPTAC or submitters.
merged_mean_median_sd_shared <- merged_mean_median_sd[,str_detect(colnames(merged_mean_median_sd), "Unshared_") == FALSE]
merged_mean_median_sd_shared_vector <- merged_mean_median_sd_shared[2,] %>% unlist()
hist(merged_mean_median_sd_shared_vector,
breaks = 1000,
main = "median value used for normalization at CPTAC or submitters",
xlab = "median",
xlim = c(-3.0, 1.0))
quantile(merged_mean_median_sd_shared_vector, probs = c(0.25, 0.5, 0.75))この時点で解析に用いている全症例のうち、どのくらいの割合で欠損値があるのかをタンパク質ごとに出す。それをベクトルpercent_naとして保存し、それをデータフレームmerged_normalized_log2ratioにcbind()で加えておく。この列は、低クオリティーのタンパク質を解析から除外するために用いる。
# code 10
# Calculate percent of NA in a row, meaning decide which genes were how many NAs are conteined.
# MARGIN = ; a vector giving the subscripts which the function will be applied over. E.g., for a matrix 1 indicates rows, 2 indicates columns, c(1, 2) indicates rows and columns. Where X has named dimnames, it can be a character vector selecting dimension names.
percent_na <- apply(merged_normalized_log2ratio, 1, function(x) {sum(is.na(x))/ncol(merged_normalized_log2ratio)})
merged_normalized_log2ratio_w_percent_na <- cbind(merged_normalized_log2ratio, percent_na)
merged_normalized_log2ratio_w_percent_na$percent_na
nrow(merged_normalized_log2ratio_w_percent_na) # 20414 genes
ncol(merged_normalized_log2ratio_w_percent_na) # 11435 = 11434 cases + percent_na code 11で、上記で計算した列percent_naの値は、どんな範囲にあるのかをヒストグラムで確認する。
# code 11
# Distribution of percent of NAs.
hist(merged_normalized_log2ratio_w_percent_na$percent_na,
breaks = 10000,
main = "Histogram of NA ratio",
xlab = "NA ratio",
xlim = c(0, 1.00))
# Distribution of percent of NAs.
hist(merged_normalized_log2ratio_w_percent_na$percent_na,
breaks = 10000,
main = "Histogram of NA ratio",
xlab = "NA ratio",
xlim = c(0.85, 1.00))
# Distribution of percent of NAs.
hist(merged_normalized_log2ratio_w_percent_na$percent_na,
breaks = 10000,
main = "Histogram of NA ratio",
xlab = "NA ratio",
xlim = c(0.0, 0.2))色々と悩んだが、NAとなっているタンパク質が、全症例のうち10%を下回る、すなわち、全症例の90%より多くの症例で検出されているタンパク質を解析する。そうすると、5208個のタンパク質が残る。
# code 12
# If a gene was detected in more than 90 % of patients, a gene was including in analysis (alternatively, a gene was detected in only 10 % of patients, a gene was omitted from analysis.)
merged_normalized_log2ratio_2 <- merged_normalized_log2ratio_w_percent_na %>% filter(percent_na < 0.1)
nrow(merged_normalized_log2ratio_2) # 5208 genes were remained.
ncol(merged_normalized_log2ratio_2) # 11435 = 11434 cases + percent_na
# Add "Gene" into column name.
merged_normalized_log2ratio_2 <- merged_normalized_log2ratio_2 %>% rownames_to_column(var = "Gene")
# Pick up genes with NA ratio > 0.1.
gene_to_be_omitted <- merged_normalized_log2ratio_w_percent_na[merged_normalized_log2ratio_w_percent_na$percent_na >= 0.1,]$Gene
length(gene_to_be_omitted) # 15206 genesそして、解析の邪魔になるので、データフレームmerged_normalized_log2ratio_2の列percent_naを削除して、新しくデータフレームmerged_normalized_log2ratio_3を作成する。
# code 13
# Transform the dataset to tibble.
merged_normalized_log2ratio_3 <- merged_normalized_log2ratio_2 %>% as_tibble()
# Remove unnecessary columns.
merged_normalized_log2ratio_3 <- merged_normalized_log2ratio_3 %>%
select(-one_of("percent_na"))
# Set column name as "Gene".
merged_normalized_log2ratio_3 <- merged_normalized_log2ratio_3 %>% column_to_rownames(var = "Gene")
ncol(merged_normalized_log2ratio_3) # 11434 cases
nrow(merged_normalized_log2ratio_3) # 5208 genesデータフレームmerged_normalized_log2ratio_3の中には、症例ではないタンパク質発現プロファイルもある。今考えてみれば、これらは意味がわからない。確かに、MSのスペクトルを改めて解析するっていうならば、それはよくわかる。その場合はQCやControlは考えられる。でもWithdrawって何だよ。そんなもん、リポジットするんじゃあねえよって感じである。そういうことで、これらは除く必要がある。それらを除いたものがデータフレームmerged_normalized_log2ratio_5である。ここはgrepl()で拾ってきた。str_detect()でも同じことが出来ると思うような気がする。出来たデータフレームの列名を見ていると、まだUnsharedのタンパク質発現プロファイルが残っているらしい。それも除いて、データフレームmerged_normalized_log2ratio_6とする。この列名は今後見直したり、使用する可能性もあるので、colnames()で列名を取ってきて、それをテキストファイルとして保存しておく。
# code 14
# This collection of the files contain QC files, which were probably used for calculating ratio of proteins for each sample at other laboratory. Therefore they will be omitted from the dataset.
merged_normalized_log2ratio_4 <-merged_normalized_log2ratio_3 %>% select(
colnames(merged_normalized_log2ratio_3)[!grepl(pattern = "^(QC|Withdrawn)", colnames(merged_normalized_log2ratio_3), ignore.case = FALSE)]
)
merged_normalized_log2ratio_5 <-merged_normalized_log2ratio_4 %>% select(
colnames(merged_normalized_log2ratio_4)[!grepl(pattern = "POOL|CONTROL", colnames(merged_normalized_log2ratio_4), ignore.case = TRUE)]
)
# merged_normalized_log2ratio_6 <-merged_normalized_log2ratio_5 %>% select(
# colnames(merged_normalized_log2ratio_5)[!grepl(pattern = "normal", colnames(merged_normalized_log2ratio_5), ignore.case = TRUE)]
# )
merged_normalized_log2ratio_6 <-merged_normalized_log2ratio_5 %>% select(
colnames(merged_normalized_log2ratio_5)[!grepl(pattern = "Unshared", colnames(merged_normalized_log2ratio_5), ignore.case = TRUE)]
)
ncol(merged_normalized_log2ratio_6) # 5144
file.remove("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/colnames_merged_normalized_log2ratio_6.txt")
cat(colnames(merged_normalized_log2ratio_6), sep = "\n", file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/colnames_merged_normalized_log2ratio_6.txt")
# 5144 cases were remained, and they will be used for further analysis.code 15で、正規化されている値を正規化前に戻す。ここではデータフレームよりもマトリックスの方が計算がしやすいと思う。なので、code 14で作成したデータフレームmerged_normalized_log2ratio_6を、as.matrix()でマトリックスmerged_normalized_log2ratio_6_matに変換する。また、正規化前の値に戻すためには、中央値が必要なので、対応する列の中央値をデータフレームmerged_mean_median_sdから取ってきて、データフレームmerged_mean_median_sd_2とし、さらにそれをマトリックスmerged_mean_median_sd_2_matに変換する。それらを使ってcode 8にある計算に従って正規化前の値に戻すわけだが、このためにsweep()を使用する。sweep()は、+とかーとかxとかの計算を演算子を行(MARGIN =1)か列(MARGIN = 2)に対してザッと行う(sweep)するための関数であり、こんな時に便利である。そうして正規化前に出来上がった値をマトリックスmerged_analysis_before_normalization_ratioとする。ここで、一応as.numeric()で全部数値になっていることを確認する。おそらくマトリックスだし、sweep()とかでエラーが出なかったから問題はないはずだが、念の為。そして、それをデータフレームmerged_analysis_before_normalization_ratio_dfに直し、colnames()を使って列名を一致させておく。
# code 15
## Transform data frame "merged_normalized_log2ratio_6" into matrix "merged_normalized_log2ratio_6_mat"
merged_normalized_log2ratio_6_mat <- as.matrix(merged_normalized_log2ratio_6)
view(merged_normalized_log2ratio_6_mat[1:100, 1:100]) # check the data.
## Retrieve the cases in data frame "merged_mean_median_sd" that are same as the cases in the matrix "merged_normalized_log2ratio_6_mat".
merged_mean_median_sd_2 <-merged_mean_median_sd[,colnames(merged_normalized_log2ratio_6_mat)]
## Transform the data frame "merged_mean_median_sd_2" into matrix "merged_mean_median_sd_2_mat"
merged_mean_median_sd_2_mat <- as.matrix(merged_mean_median_sd_2)
view(merged_mean_median_sd_2_mat[, 1:100]) # check the data.
## the matrix "merged_normalized_log2ratio_6_mat" was already centered with median value. To get back to the value before normalization, the median value was added into each column. The matrix "merged_log2ratio_6_mat" was the data before normalization, but it is still log2 transformed value.
merged_log2ratio_6_mat <- sweep(x = merged_normalized_log2ratio_6_mat,
MARGIN = 2,
STATS = merged_mean_median_sd_2_mat[2,],
FUN = "+") # The matrix "merged_log2ratio_6_mat" is normalized_log2ratio + median = log2ratio
## the matrix "merged_analysis_before_normalization_ratio" was argument of the data.
merged_analysis_before_normalization_ratio <- 2^merged_log2ratio_6_mat # the matrix "merged_analysis_before_normalization_ratio" is raw ratio before normalization. If the value gets back to normalized ratio, following equation should be used.
# log2(2^merged_log2ratio_6_mat) - median = normalized_log2ratio
view(merged_analysis_before_normalization_ratio[1:100, 1:100]) # check the data.
## make sure the value of the matrix "merged_analysis_before_normalization_ratio" was numeric.
merged_analysis_before_normalization_ratio <- apply(merged_analysis_before_normalization_ratio, 2, as.numeric)
## make sure the rownames of the matrix "merged_analysis_before_normalization_ratio" is same as that of the matrix "merged_log2ratio_6_mat", which is original dataset.
rownames(merged_analysis_before_normalization_ratio) <- rownames(merged_log2ratio_6_mat)
view(merged_analysis_before_normalization_ratio[1:100, 1:100]) # check the data.
## Get the matrix "merged_analysis_before_normalization_ratio" back to data frame "merged_analysis_before_normalization_ratio_df"
merged_analysis_before_normalization_ratio_df <- data.frame(merged_analysis_before_normalization_ratio)
colnames(merged_analysis_before_normalization_ratio_df) <- colnames(merged_normalized_log2ratio_6)
#
# merged_analysis_before_normalization_ratio_df[is.na(merged_analysis_before_normalization_ratio_df)] <- 0
#
# options(scipen = 100)これらは今後解析に用いていく症例であり、もしかしたらどこかで参照する可能性もあるため、列名をベクトルsamples_to_be_analyzedとして保存する。
# code 16
# "merged_analysis_before_normalization_ratio_df" is the dataset that was put the normalized dataset back to the (possiblly) original dataset.
# "samples_to_be_analyzed" at following line was column names, which are file names of tmt10, in dataset "merged_analysis_before_normalization_ratio_df".
# Files in "samples_to_be_analyzed" will be used for further analysis, such as calculation of immune suppression score.
view(colnames(merged_analysis_before_normalization_ratio_df))
samples_to_be_analyzed <- colnames(merged_analysis_before_normalization_ratio_df)
# In case of file will be saved on C drive.
file.remove("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/samples_to_be_analyzed.txt")
cat(samples_to_be_analyzed, sep = "\n", file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/samples_to_be_analyzed.txt")データフレームmerged_analysis_before_normalization_ratio_dfは、正規化前のタンパク質発現プロファイルであるであり、その列名(症例)と行名(タンパク質)と、他のデータ、例えば、正規化後のタンパク質発現プロファイル(データフレームmerged_normalized_log2ratio_6)と症例やタンパク質は一致させなければならない。ここでは、code 16で準備したベクトルsamples_to_be_analyzed(今後使用することになるだろう症例)と%in%を使うことで症例とタンパク質を合わせ、それらが合っているかどうか確認をする。タンパク質はpercent_naでフィルターして以来触っていないので、合っているはずである。なので、症例数を合わせれば良いはず。
# code 17
## the data frame "merged_normalized_log2ratio_6" was the dataframe without unnecessary columns. This data will be used for further analysis. Here, column names (which are cases) and column number (which is number of cases) will be checked by colnames(), ncol(), and length().
colnames(merged_normalized_log2ratio_6)
ncol(merged_normalized_log2ratio_6) # 5144
samples_to_be_analyzed
length(samples_to_be_analyzed) # 5144
## Check if the case number between dataframe "merged_normalized_log2ratio_6" and "samples_to_be_analyzed" is same or not.
table(!colnames(merged_normalized_log2ratio_6) %in% samples_to_be_analyzed) # FALSE 5144 means that all column if the data frame "merged_normalized_log2ratio_6" is same as the vectior "samples_to_be_analyzed".
check_match <- colnames(merged_normalized_log2ratio_6)[colnames(merged_normalized_log2ratio_6) %in% samples_to_be_analyzed]
check_unmatch <- colnames(merged_normalized_log2ratio_6)[!colnames(merged_normalized_log2ratio_6) %in% samples_to_be_analyzed]
length(check_match) # 5144
length(check_unmatch) # 0
length(check_match) + length(check_unmatch) # 5144
view(colnames(merged_normalized_log2ratio_6))
view(samples_to_be_analyzed)
view(check_unmatch)
## the case number between dataframe "merged_normalized_log2ratio_6" and "samples_to_be_analyzed" is same.データフレームmerged_normalized_log2ratio_6の列名・列数はベクトルsamples_to_be_analyzedと合っていそうなので、それらをmissForestを使うためのフォーマットに変換しておく。データフレームmerged_analysis_before_normalization_ratio_df、マトリックスmerged_normalized_log2ratio_6_matをt()で転置させ、それぞれマトリックスmerged_analysis_before_normalization_ratio_mat_t、マトリックスmerged_normalized_log2ratio_6_mat_tを作る。ここで、それぞれの列名(タンパク質発現プロファイル名)と行名(各タンパク質)は変換前の物を当てておく。転置しているので、もとのデータフレームの行名もしくは列名が転置後の列名もしくは行名になるはず。事前にcolnames()とrownames()で取ってきたものを、それぞれ当てれば良い。
このマトリックスmerged_analysis_before_normalization_ratio_mat_t、マトリックスmerged_normalized_log2ratio_6_mat_t、マトリックスmerged_log2ratio_6_matはmissForestに使用する。マトリックスmerged_log2ratio_6_matはcode 15で作成してある。
なぜマトリクス3つを使用するかといえば、どれが一番良いのか分からなかったためである。missForestによって欠損値をimputationされたデータを使って中央値を出してみて、それがCPTACで出された中央値に一番近いものを解析で使用していこうと思う。
| マトリックス名 | 内容 | 行 | 列 |
|---|---|---|---|
| merged_analysis_before_normalization_ratio_mat_t | 正規化前 真数 | タンパク質 | ファイル名 |
| merged_log2ratio_6_mat_t | 正規化前 対数(底2) | タンパク質 | ファイル名 |
| merged_normalized_log2ratio_6_mat_t | 正規化後 対数(底2) | タンパク質 | ファイル名 |
# code 18
## prepare the dataset that is argument before normalization.
cases_pre_argu_normalization <- colnames(merged_analysis_before_normalization_ratio_df)
proteins_pre_argu_normalization <- rownames(merged_analysis_before_normalization_ratio_df)
length(cases_pre_argu_normalization) # 5144
length(proteins_pre_argu_normalization) # 5208
merged_analysis_before_normalization_ratio_mat <- as.matrix(merged_analysis_before_normalization_ratio_df)
merged_analysis_before_normalization_ratio_mat_t <- t(merged_analysis_before_normalization_ratio_mat)
view(merged_analysis_before_normalization_ratio_mat_t[1:1000, 1:10]) # check the data.
## prepare the dataset before normalization, it is still logarithm.
cases_pre_normalization_log <- colnames(merged_log2ratio_6_mat)
proteins_pre_normalization_log <- rownames(merged_log2ratio_6_mat)
length(cases_pre_normalization_log) # 5144
length(proteins_pre_normalization_log) # 5208
merged_log2ratio_6_mat_t <- t(merged_log2ratio_6_mat)
view(merged_log2ratio_6_mat_t[1:1000, 1:10]) # check the data.
## prepare the dataset that is already normalized by CPTAC or submitters.
cases_post_normalization <- colnames(merged_normalized_log2ratio_6_mat)
proteins_post_normalization <- rownames(merged_normalized_log2ratio_6_mat)
length(cases_post_normalization) # 5144
length(proteins_post_normalization) # 5208
merged_normalized_log2ratio_6_mat_t <- t(merged_normalized_log2ratio_6_mat)
view(merged_normalized_log2ratio_6_mat_t[1:1000, 1:10]) # check the data.
## check cases and proteins
table(cases_pre_argu_normalization %in% cases_pre_normalization_log) # TRUE 5144
table(proteins_pre_argu_normalization %in% proteins_pre_normalization_log) # TRUE 5208
table(cases_pre_argu_normalization %in% cases_post_normalization) # TRUE 5144
table(proteins_pre_argu_normalization %in% proteins_post_normalization) # TRUE 5208
table(cases_pre_normalization_log %in% cases_post_normalization) # TRUE 5144
table(proteins_pre_normalization_log %in% proteins_post_normalization) # TRUE 5208
ここまでの結果をRDataとして保存しておく。
# code 19
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_1.RData")missForestによる欠損値の補完(imputation)
missForestは、参照するデータセットなしにランダムフォレストを行うことで欠損値の補完を行うパッケージである。欠損値として最初に何かしらの値を代入し、そこを欠損値を含んでいない列を使ってランダムフォレストによって逐次予測していき、直前の予測値と次の予測値の差に変化がなくるまでそれを繰り返す、ということをやっているらしい。
ゼロで欠損値を補完するのが駄目な理由
まず、以下のヒストグラムを見てみる。これは、code 8で、データフレームmerged_normalized_log2ratioの一列目を取ってきて作成したベクトルnormalized_log2ratioのヒストグラムで、欠損値はそのままにしてhist()を実行した結果である。まぁ、こんな感じかってところである。
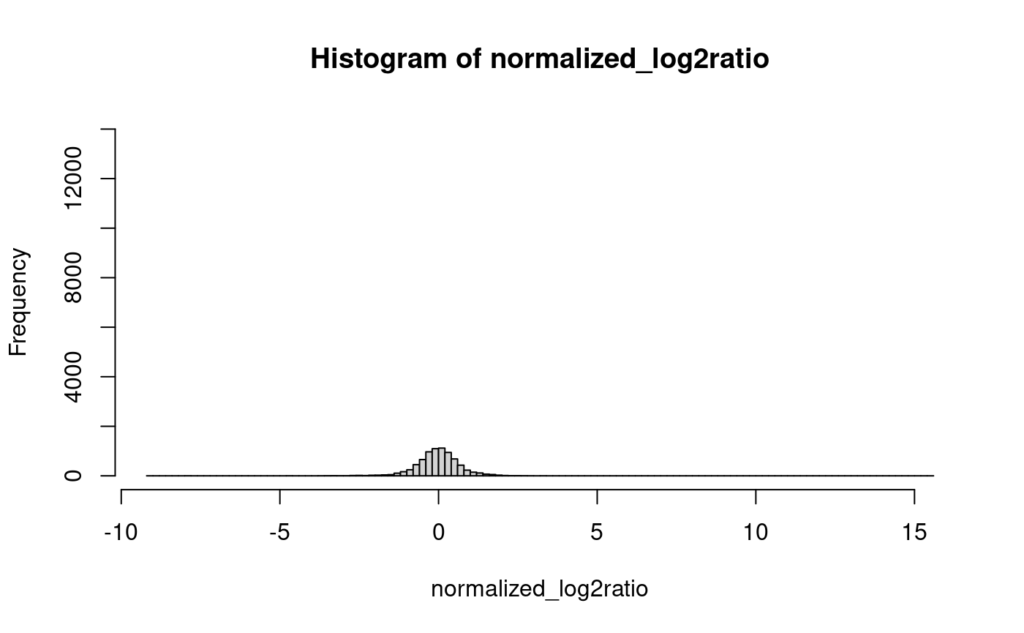
そして、以下が欠損値にゼロを入れてhist()を実行した結果である。
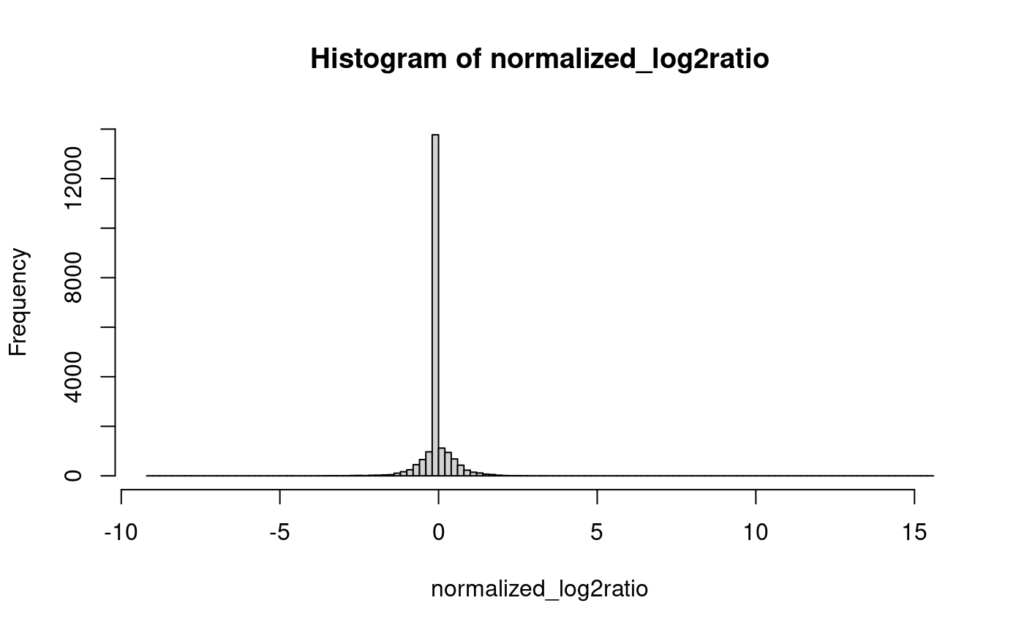
これだと、なんか駄目な気がする。なんか凄い分布になってしまった。このようなデータでは何かしらの統計分布にフィッティングして解析するようなことが出来ないことがわかる。なので、欠損値ではない値を参考にして、それとの差を最小にするようにランダムフォレストを使って欠損値を埋めるように計算するのがmissForestである。他にも、PCA(Principal Component Analysis; 主成分分析)だったりkNN(k-Nearest Neihbers; k近傍法)などを使って似た値で埋めていく方法もあるらしいが、ゼロのように単一の値で補完するのは良くないって言ってるんだから、PCAもkNNもあまり良くないように思う。注意すべきなのが、あまりにも沢山欠損値があるデータはmissForestで補完したところで計算時間が増えたり、観測値と似た値を代入できなかったりするらしいので、欠損値はなるべく少ないようにした方が良いのだろう。それに、そもそも、欠損値が多すぎるタンパク質は解析から除いたほうが良い。そのようなタンパク質が数例のみで観察され、他の90%の症例では検出出来ないような場合、もはやそのタンパク質はそもそも検出が難しくて検出出来なかったりする物だろうから、検出されている物は信頼できるのかどうかわからないし、その他の90%は欠損値を補完して得られた値で比較することになり、その比較には意味があるのか疑問である。なのでこの解析では、全症例のうち欠損値が10%より低いタンパク質を使用している。これはcode 12にある。
上記が一般的な説明になると思う。しかし、これは本当だろうか。前述したが、だとしたらなぜCPTACは欠損値を埋めるのにゼロを使っているのだろうか(欠損値をゼロで埋めると、元からある中央値や平均値を再現できるため)。これは、用途によってはゼロも使用可能であるってことではないのだろうか。ゼロで補完した場合は何かしらの統計分布、例えば、RNA-seqで言うところの負の二項分布などにフィッティングをしなかったら良いだけではないか。それに、フィッティングするにしても、もしかして欠損値は除いて全体で計算し、その結果を使って後から欠損値を予測・推定すれば、それで欠損値の補完になるのではないだろうか….むしろ、こちらの方がデータ全体の統計的分布に従うような値で欠損値を補完できると思うのだが。これだと結局missForestみたいな方法で計算する羽目にはなるか….でもmissForestだと統計的分布は、欠損値ではない他の値の統計分布に従うような気がする。知らんけど。
欠損値の補完は正直、何が最適なのかよくわからない。結局のところ、これは推測値であり実測値ではない。
missForestの実行
まず、上述の通りに3つのデータセットに対してmissForestを適用していく。missForestの計算にはかなりの時間を要するので、ぜひ並列化した方がよい。ここでは、マニュアルに従ってdoParallelを使っていく。一つの計算が終わったら、おまじないとしてgc(T,T,T)でメモリをクリアしておく。missForestは、5000行x5000列くらいのマトリックスで計算に1.2日くらいは必要である。
まずはマトリックスmerged_analysis_before_normalization_ratio_mat_tである。これはCPTACによる正規化前の値で、真数に直してある。
# code 20
# Impute missing values using missForest for dataset BEFORE normalization
cl <- makeCluster(detectCores())
registerDoParallel(cl)
start_time_pre_argu <- Sys.time()
set.seed(20260121) # for reproducibility
result_missForest_pre_argu <- missForest(merged_analysis_before_normalization_ratio_mat_t, parallelize = "variables", verbose = TRUE)
end_time_pre_argu <- Sys.time()
stopCluster(cl)
gc(T,T,T)
gc(T,T,T)
spendintg_time_pre_argu <- end_time_pre_argu - start_time_pre_argu
spendintg_time_pre_argu # Time difference of 20.40168 hours
# 15.24111 hours will be spent for missForest with NH-U12A.次にマトリックスmerged_log2ratio_6_mat_tをmissForestする。これは正規化前の値で、値としては底2の対数である。
# code 21
# Impute missing values using missForest for dataset AFTER normalization
# The matrix "merged_normalized_log2ratio_6_mat_t" is from the matrix "merged_normalized_log2ratio_6_mat".
# The matrix "merged_normalized_log2ratio_6_mat" is normalized_log2ratio (median-normalized log2ratio) + median = log2ratio. log2ratio is the value before log2 transform.
cl <- makeCluster(detectCores())
registerDoParallel(cl)
start_time_pre <- Sys.time()
set.seed(20260121) # for reproducibility
result_missForest_pre <- missForest(merged_log2ratio_6_mat_t, parallelize = "variables", verbose = TRUE)
end_time_pre <- Sys.time()
stopCluster(cl)
gc(T,T,T)
gc(T,T,T)
spendintg_time_pre <- end_time_pre - start_time_pre # Time difference of 1.196548 days
spendintg_time_pre # Time difference of 1.217632 days次に、CPTACで正規化されたデータをmissForestする。正規化された値であり、かつ、底2の対数である。
# code 22
# Impute missing values using missForest for dataset AFTER normalization
# The matrix "merged_normalized_log2ratio_6_mat_t" is from the matrix "merged_normalized_log2ratio_6_mat".
# The matrix "merged_normalized_log2ratio_6_mat" is normalized_log2ratio (median-normalized log2ratio) + median = log2ratio. log2ratio is the value before log2 transform.
cl <- makeCluster(detectCores())
registerDoParallel(cl)
start_time_post <- Sys.time()
set.seed(20260121) # for reproducibility
result_missForest_post <- missForest(merged_normalized_log2ratio_6_mat_t, parallelize = "variables", verbose = TRUE)
end_time_post <- Sys.time()
stopCluster(cl)
spendintg_time_post <- end_time_post - start_time_post
spendintg_time_post # Time difference of 1.129523 days
gc(T,T,T)
gc(T,T,T)いちいちこんな長い計算をするのは嫌なので、終わったらRDataとして保存する。
# code 23
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_2.RData")3つのうちどのデータを使うか
上述したように、正規化前後のデータを使うのか、対数と真数どちらが良いのか、いまいちよくわからない。なので、ここからはどのデータが一番良いのかを判断するために、色々とデータを見ていこうと思う。
ヒストグラムを表示してみる
とりあえず、得られた結果をヒストグラムで表示して、各々のデータがどんな分布なのか確認してみる。missForestの結果はリストの要素ximpに入っているので、それをhist()に渡す。
…駄目だ。結局わからん。どれも一緒の結果に見える。ヒストグラムだけではどのデータを用いたものが良いのか、わからなかった。
# code 24
# check histogram
hist(merged_analysis_before_normalization_ratio_mat_t, breaks = 1000) # , ylim = c(0, 14000)
hist(result_missForest_pre_argu$ximp[1:100, 1:100], breaks = 1000)
hist(merged_analysis_before_normalization_ratio_mat_t[1:100, 1:100], breaks = 1000, xlim = c(0, 4))
hist(result_missForest_pre_argu$ximp[1:100, 1:100], breaks = 1000, xlim = c(0, 4))
hist(merged_log2ratio_6_mat_t[1:100, 1:100], breaks = 1000)
hist(result_missForest_pre$ximp[1:100, 1:100], breaks = 1000)
hist(merged_normalized_log2ratio_6_mat_t[1:100, 1:100], breaks = 1000)
hist(result_missForest_post$ximp[1:100, 1:100], breaks = 1000)欠損値(NA)の場所をmissForest前後で取得してみる
次に、もはやどうやって評価するか迷ってしまっているので、ひとまずマトリックスでNAだった場所を取ってくる。これはwhich(is.na(), , arr.ind = TRUE)で出来る。取ってきたNAの場所は、それぞれベクトルNA_location_pre_argu、NA_location_pre、NA_location_postとした。これだけでは、特に何もわからんかった。
# code 25
# merged_analysis_before_normalization_ratio_df # data frame, for "result_missForest_pre_argu" object
# merged_log2ratio_6_mat # matrix, for "result_missForest_pre" object
# merged_normalized_log2ratio_6 # data frame, for "result_missForest_post" object
## transform matrix "merged_log2ratio_6_mat" to data frame
merged_log2ratio_6_df <- data.frame(merged_log2ratio_6_mat)
## Check the dimension of each data frame. They had same dimension.
dim(merged_log2ratio_6_df) # 5208 5202
dim(merged_normalized_log2ratio_6) # 5208 5202
dim(merged_analysis_before_normalization_ratio_df) # 5208 5202
## Retrieve location of NA value in the dataframe "merged_normalized_log2ratio_6" (which is normalized intensity at CPTAC or submitter) and "merged_analysis_before_normalization_ratio_df"(which is the intensity before normalization).
NA_location_pre_argu <- which(is.na(merged_analysis_before_normalization_ratio_df), arr.ind = TRUE) %>% data.frame()
NA_location_pre <- which(is.na(merged_log2ratio_6_df), arr.ind = TRUE) %>% data.frame()
NA_location_post <- which(is.na(merged_normalized_log2ratio_6), arr.ind = TRUE) %>% data.frame()
# check if the location of NA of dataframe "merged_normalized_log2ratio_6" and the dataframe "merged_analysis_before_normalization_ratio_df" is same or not. The results showed all location for both dataframe was same.
#
table(NA_location_pre$col %in% NA_location_pre_argu$col) # TRUE 748682, All true
table(NA_location_pre$row %in% NA_location_pre_argu$row) # TRUE 748682, All true
#
table(NA_location_pre_argu$col %in% NA_location_post$col) # TRUE 748682, All true
table(NA_location_pre_argu$row %in% NA_location_post$row) # TRUE 748682, All true
#
table(NA_location_pre$col %in% NA_location_post$col) # TRUE 748682, All true
table(NA_location_pre$row %in% NA_location_post$row) # TRUE 748682, All true結局わからんかったのでデータを眺めてみる
どうやったら一番良さげなmissForestの結果を選べるだろうか。全然わからなかったので、欠損値補完前後のデータをview()で眺めておく。
# code 26
# Check what the data looks like.
view(merged_analysis_before_normalization_ratio_mat_t[1:118, 1:118]) # I do not know why I showed 1:118, but the area contained NA values.
view(result_missForest_pre_argu$ximp[1:100, 1:100])# missForest results of dataset before normalization and back to arguments
view(merged_log2ratio_6_mat_t[1:100, 1:100])
view(result_missForest_pre$ximp[1:100, 1:100]) # missForest results of dataset before normalizatiom.
view(merged_normalized_log2ratio_6_mat_t[1:100, 1:100])
view(result_missForest_post$ximp[1:100, 1:100]) # missForest results of normalized dataset データフレームに直してみる
そういえば、マトリックスresult_missForest_pre_argu、result_missForest_pre、result_missForest_postをデータフレームに直すことにする。このとき、データをt()で転置して、列をタンパク質発現プロファイル名に、行を各タンパク質に直しておく。ここで注意点がある。ここのcode 27で作成するデータフレームresult_missForest_pre_argu_dfだが、次のcode 28で作成するデータフレームmerged_mean_median_sd_3で対象とする症例を取ってくるのに使用するだけである。特にこのデータフレームresult_missForest_pre_argu_dfを他の計算に使用する、とかではない。我ながら紛らわしい。ただし、ここをやっているとき、どうやってmissForestの結果を選んでくるか迷走中だったので、こういうことをしてしまったのだろう。紛らわしいのであまり良いことではない。code 28で作成するデータフレームresult_missForest_pre_argu_log2_dfを、その後の解析に使用する。
# code 27
# Results of missForest were matrix. Transform the matrix into dataframe. Following is for the intensity before normalization ("result_missForest_pre"). The column and the row should respectively be cases and peptids and therefore it transposed by t().
# # cases_pre_argu_normalization
# # proteins_pre_argu_normalization
# #
# # cases_pre_normalization_log
# # proteins_pre_normalization_log
# #
# # cases_post_normalization
# # proteins_post_normalization
result_missForest_pre_argu_df <- t(result_missForest_pre_argu$ximp) %>% data.frame()
colnames(result_missForest_pre_argu_df) <- cases_pre_argu_normalizationmissForestの結果をlog2変換した正規化前の値に直す
上記code 27で作成したデータフレームresult_missForest_pre_argu_dfに対応する中央値を抽出して、マトリックスmerged_mean_median_sd_3_matを作成する。
未だにどうやってmissForestの結果をどうやって選ぶか迷っていたので、このcode 28で「まずは一旦全部同じような形式に直そうか。」とかになったんだと思う。ここから、各データを「正規化前のlog2変換(底2の対数)された値」に合わせたデータフレームを作成していく。まずは正規化前の真数のデータresult_missForest_pre_arguからである。これだけlog2()変換する必要がある。t()で転置して列名をタンパク質発現プロファイル名、行名をタンパク質に直した上で、それをlog2()に入れて底2の変数に変換し、マトリックスresult_missForest_pre_argu_log2を作成する。そして、このマトリックスを念のためにapply(MAGIN =2, as.numeric)で数字にし、最後にこれをデータフレームに変換し、データフレームresult_missForest_pre_argu_log2_dfを作成する。ここの最後に、各データを使ったmissForestの結果の行と列が、全部同じかどうかを確認しておいた。
# code 28
# Check if the median value of "result_missForest_pre_df" is closer to the median value that is calculated by CPTAC or submitters.
## At first, check data.
view(result_missForest_pre_argu_df[1:200, 1:200]) # check data. This is data before normalization that was calculated with median.
view(merged_mean_median_sd_2[1:3, 1:200]) # check data. This is mean, median and sd value that was used for median normalization.
view(merged_normalized_log2ratio_6_mat[1:200, 1:200]) # check data. This is normalized intensity by CPTAC or submitters.
## Match the cases in data frame "merged_mean_median_sd_2" with the cases in data frame "result_missForest_pre_df".
merged_mean_median_sd_3 <- merged_mean_median_sd_2 %>% dplyr::select(all_of(colnames(result_missForest_pre_argu_df)))
merged_mean_median_sd_3_mat <- as.matrix(merged_mean_median_sd_3)
view(merged_mean_median_sd_3_mat[1:3, 1:100]) # check data
view(t(result_missForest_pre_argu$ximp)[1:100, 1:100]) # check data
## log2 transform of missForest result before normalization.
result_missForest_pre_argu_log2 <- log2(t(result_missForest_pre_argu$ximp))
view(result_missForest_pre_argu_log2)[1:10, 1:10]# check data
## Normalization again. Later, check if the estimated NA value is close to median value by CPTAC or submitter.
## Remind; log2(merged_normalized_log2ratio_6_mat) - median = normalized_log2ratio
# result_missForest_pre_argu_df_median_normalized <- sweep(x = result_missForest_pre_argu_log2,
# MARGIN = 2,
# STATS = merged_mean_median_sd_3_mat[2,],
# FUN = "-")
# view(result_missForest_pre_argu_df_median_normalized[1:100, 1:100]) # check data
## Make sure the value of data frame "result_missForest_pre_df_median_normalization" is numeric.
# result_missForest_pre_argu_df_median_normalized <- apply(result_missForest_pre_argu_df_median_normalized, 2, as.numeric)
result_missForest_pre_argu_log2 <- apply(result_missForest_pre_argu_log2, 2, as.numeric)
## Then the matrix "result_missForest_pre_df_median_normalization" transformed to data frame "result_missForest_pre_df_median_normalization_df"
# result_missForest_pre_argu_df_median_normalized_df <- data.frame(result_missForest_pre_argu_df_median_normalized)
result_missForest_pre_argu_log2_df <- data.frame(result_missForest_pre_argu_log2)
## Retrieve column name (case) and row name(protein).
colnames(result_missForest_pre_argu_log2_df) <- cases_pre_argu_normalization
rownames(result_missForest_pre_argu_log2_df) <- proteins_pre_argu_normalization
# # cases_pre_argu_normalization
# # proteins_pre_argu_normalization
# #
# # cases_pre_normalization_log
# # proteins_pre_normalization_log
# #
# # cases_post_normalization
# # proteins_post_normalization
table(cases_pre_argu_normalization %in% cases_pre_normalization_log) # TRUE 5202
table(proteins_pre_argu_normalization %in% proteins_pre_normalization_log) # TRUE 5208
table(cases_pre_argu_normalization %in% cases_post_normalization) # TRUE 5202
table(proteins_pre_argu_normalization %in% proteins_post_normalization) # TRUE 5208
table(cases_pre_normalization_log %in% cases_post_normalization) # TRUE 5202
table(proteins_pre_normalization_log %in% proteins_post_normalization) # TRUE 5208code 29では正規化前の値で、底2の対数をmissForestしたデータをデータフレームに直す。これはすでに底2の対数なので、単に転置してからデータフレームに直すだけである。ここでデータフレームresult_missForest_pre_t_dfを作成する。この記事を書いていてツッコミを入れてしまったが「apply()でas.numeric()せんのかい」って感じである。最後に行例が他のデータと同じかどうか確認している。
# code 29
# merged_log2ratio_6_mat_t
result_missForest_pre_df <- t(result_missForest_pre$ximp) %>% data.frame()
colnames(result_missForest_pre_df) <- cases_pre_normalization_log
# merged_mean_median_sd_4 <- merged_mean_median_sd_2 %>% dplyr::select(all_of(colnames(result_missForest_pre_df)))
# merged_mean_median_sd_4_mat <- as.matrix(merged_mean_median_sd_4)
result_missForest_pre_t <- t(result_missForest_pre$ximp)
view(result_missForest_pre_t[1:10, 1:10])# check data
## Normalization again. Later, check if the estimated NA value is close to median value by CPTAC or submitter.
## Remind; log2(merged_normalized_log2ratio_6_mat) - median = normalized_log2ratio
# result_missForest_pre_df_median_normalized <- sweep(x = result_missForest_pre_t,
# MARGIN = 2,
# STATS = merged_mean_median_sd_4_mat[2,],
# FUN = "-")
result_missForest_pre_t_df <- data.frame(result_missForest_pre_t)
colnames(result_missForest_pre_t_df) <- cases_pre_normalization_log
view(result_missForest_pre_t_df[1:100, 1:100])
view(cases_pre_normalization_log[1:100])
table(colnames(result_missForest_pre_t_df) %in% cases_pre_normalization_log) # TRUE 5144
table(colnames(result_missForest_pre_t_df) %in% cases_pre_normalization_log) # TRUE 5144
table(colnames(result_missForest_pre_df) %in% cases_pre_normalization_log) # TRUE 5144
# colnames(result_missForest_pre_df_median_normalized_df) <- cases_pre_normalization_log
# rownames(result_missForest_pre_df_median_normalized_df) <- proteins_pre_normalization_log
# # cases_pre_argu_normalization
# # proteins_pre_argu_normalization
# #
# # cases_pre_normalization_log
# # proteins_pre_normalization_log
# #
# # cases_post_normalization
# # proteins_post_normalization今度は、CPTACで正規化された値(底2の対数)をデータフレームに直す。これは底2の対数であるが、これは未だ正規化後の値なので、正規化前に直す必要がある。そのために、まずはmerged_mean_median_sd_2から一致するファイル名を抽出してマトリックスmerged_mean_median_sd_4_matを作成し、そのマトリックスの2行目の値、すなわち中央値とsweep()を使って、正規化前の値に直しておく。これは底2の対数のままで良い。
これ、記事を書いていてわかったのだが、result_missForest_post_dfというオブジェクトはマトリックスのままになっている…なんてこった。ここでデータフレームに直してないことに、ここで気がついた。
# code 30
## matched the dimension (I mean, they are columns and rows) of "result_missForest_post_df" with data frame "result_missForest_pre_df_median_normalization", and then transformed it to data frame "result_missForest_post_df"
view(t(result_missForest_post$ximp)[1:100, 1:100]) # check data
merged_mean_median_sd_4 <- merged_mean_median_sd_2 %>% dplyr::select(all_of(colnames(t(result_missForest_post$ximp))))
merged_mean_median_sd_4_mat <- as.matrix(merged_mean_median_sd_4)
table(colnames(t(result_missForest_post$ximp)) %in% colnames(merged_mean_median_sd_4)) # TRUE 5144
result_missForest_post_df <- sweep(x = t(result_missForest_post$ximp),
MARGIN = 2,
STATS = merged_mean_median_sd_4_mat[2,],
FUN = "+")
## Retrieve column name for futher analysis.
colnames(result_missForest_post_df) <- cases_post_normalization欠損値に代入された値を取得する
code 31では、code 25で作成したデータフレームNA_location_postに、mutate()で列pre_argu、列pre、列postを付け加え、データフレームcheck_after_missForestを作成する。列pre_argu、列pre、列postに、missForestで補完したときの値、すなわち、元々欠損値だったところに、missForestによって代入された値を入れる。この処理であるが、計算自体はどうやら18分程度で終わっているが、データをメモリに入れるのに非常に時間がかかっていた。数時間かかったんじゃあないだろうか。これ本当にRの駄目なところである。
# code 31
## check dimension of data frame.
dim(result_missForest_pre_argu_log2_df) # [1] 5208 5144 # This is centered with median value.
dim(result_missForest_pre_t_df) # [1] 5208 5144 # This is centered with median value.
dim(result_missForest_post_df) # [1] 5208 5144 # This is centered with median value at CPTAC or submitters.
## check if columns (cases) and rows (protein) between data frame "result_missForest_pre_argu_df_median_normalized_df" (intensity normalized by median value after imputation with missForest) and "result_missForest_post_df" (intensity of normalized by CPTAC or submitters and then missForest imputation)
table(colnames(result_missForest_pre_argu_log2_df) %in% colnames(result_missForest_pre_t_df)) # TRUE 5144
table(colnames(result_missForest_pre_argu_log2_df) %in% colnames(result_missForest_post_df)) # TRUE 5144
table(colnames(result_missForest_pre_t_df) %in% colnames(result_missForest_post_df)) # TRUE 5144
table(rownames(result_missForest_pre_argu_log2_df) %in% rownames(result_missForest_pre_t_df)) # TRUE 5208
table(rownames(result_missForest_pre_argu_log2_df) %in% rownames(result_missForest_post_df)) # TRUE 5208
table(rownames(result_missForest_pre_t_df) %in% rownames(result_missForest_post_df)) # TRUE 5208
## Prepare empty data frame for comparison of missForest results.
### column "pre" is for median value of data frame "result_missForest_pre_argu_df_median_normalized_df".
### column "post" is for median value of data frame "result_missForest_post_df".
check_after_missForest <- NA_location_post %>% mutate(
pre_argu = NA,
pre = NA,
post = NA
)
# Put median value for each NA location in data frame "result_missForest_pre_argu_df_median_normalized_df" and "result_missForest_post_df" into column "pre" and "post" in data frame "check_after_missForest", respectively.
start_check_after_missForest <- Sys.time()
for(i in 1:nrow(NA_location_post)){
## for(i in 1:1000){ ## this line is just a test with small number of rows.
print(paste0("Processing row ", i, " of ", nrow(NA_location_post)))
check_after_missForest$pre_argu[i] <- result_missForest_pre_argu_log2_df[NA_location_post$row[i], NA_location_post$col[i]]
check_after_missForest$pre[i] <- result_missForest_pre_t_df[NA_location_post$row[i], NA_location_post$col[i]]
check_after_missForest$post[i] <- result_missForest_post_df[NA_location_post$row[i], NA_location_post$col[i]]
}
end_check_after_missForest <- Sys.time()
time_check_after_missForest <- end_check_after_missForest - start_check_after_missForest
time_check_after_missForest # Time difference of 18.79237 minsもともと欠損値だったところにmissForestにより代入された値の統計値を色々と見てみるが、結局、統計値をざっと見ても、正規化前の値を真数に戻した値、正規化前のlog2変換した値、正規化後のlog2変換(つまりCPTACの値そのまま)のどのデータを使ってmissForestで補完するのが、CPTACで出している中央値に一番近いのかは判断できなかった。
また、hist()の出力結果をデータフレームhist_check_after_missForest_pre_arguに入れて、どのような階層にある値が一番多いのか、とかも見てみた。しかしながら、結局どれが一番良いものなのか、判断は出来なかった。
# code 32
## median and modal value of imputed NA value. modal value was calculated by modal() in raster package.
### median value of imputed NA value
median(check_after_missForest$pre_argu) # -0.124715
median(check_after_missForest$pre) # -0.2962415
median(check_after_missForest$post) # -0.2952342
### mean value of imputed NA value
mean(check_after_missForest$pre_argu) # -0.1505831
mean(check_after_missForest$pre) # -0.3874793
mean(check_after_missForest$post) # -0.3849859
## range of the imputed NA value
quantile(check_after_missForest$pre_argu, probs = c(0.25, 0.5, 0.75)) # -0.5652116 -0.1247150 0.2215660
quantile(check_after_missForest$pre, probs = c(0.25, 0.5, 0.75)) # -0.76488152 -0.29624152 0.05727549
quantile(check_after_missForest$post, probs = c(0.25, 0.5, 0.75)) # -0.76853020 -0.29523424 0.05929829
# IQR
IQR(check_after_missForest$pre_argu) # 0.7867776
IQR(check_after_missForest$pre) # 0.822157
IQR(check_after_missForest$post) # 0.8278285
## SD
sd(check_after_missForest$pre_argu) # 0.9131712
sd(check_after_missForest$pre) # 0.7586585
sd(check_after_missForest$post) # 0.8080771
## %CV
sd(check_after_missForest$pre_argu)/mean(check_after_missForest$pre_argu) # -6.064233 # mean value is below 0.
sd(check_after_missForest$pre)/mean(check_after_missForest$pre) # -1.957933 # mean value is below 0.
sd(check_after_missForest$post)/mean(check_after_missForest$post) # -2.098979 # mean value is below 0.
## range
range(check_after_missForest$pre_argu) # -6.474371 25.291331
max(check_after_missForest$pre_argu) - min(check_after_missForest$pre_argu) # 31.7657
range(check_after_missForest$pre) # -11.205002 8.976766
max(check_after_missForest$pre) - min(check_after_missForest$pre) # 20.18177
range(check_after_missForest$post) # -8.107361 9.021220
max(check_after_missForest$post) - min(check_after_missForest$post) # 17.12858
## histogram
hist_check_after_missForest_pre_argu <- hist(check_after_missForest$pre_argu, breaks = 10000)
hist_check_after_missForest_pre <- hist(check_after_missForest$pre, breaks = 10000)
hist_check_after_missForest_post <- hist(check_after_missForest$post, breaks = 10000)
hist_check_after_missForest_pre_argu_df <- data.frame(mids = hist_check_after_missForest_pre_argu$mids, counts = hist_check_after_missForest_pre_argu$counts)
hist_check_after_missForest_pre_df <- data.frame(mids = hist_check_after_missForest_pre$mids, counts = hist_check_after_missForest_pre$counts)
hist_check_after_missForest_post_df <- data.frame(mids = hist_check_after_missForest_post$mids, counts = hist_check_after_missForest_post$counts)
hist_check_after_missForest_pre_argu_df[hist_check_after_missForest_pre_argu_df$counts == max(hist_check_after_missForest_pre_argu_df$counts), ]$counts # 3551
hist_check_after_missForest_pre_argu_df[hist_check_after_missForest_pre_argu_df$counts == max(hist_check_after_missForest_pre_argu_df$counts), ]$mids # -0.0075
hist_check_after_missForest_pre_df[hist_check_after_missForest_pre_df$counts == max(hist_check_after_missForest_pre_df$counts), ]$counts # 1425
hist_check_after_missForest_pre_df[hist_check_after_missForest_pre_df$counts == max(hist_check_after_missForest_pre_df$counts), ]$mids # -0.005
hist_check_after_missForest_post_df[hist_check_after_missForest_post_df$counts == max(hist_check_after_missForest_post_df$counts), ]$counts # 1463
hist_check_after_missForest_post_df[hist_check_after_missForest_post_df$counts == max(hist_check_after_missForest_post_df$counts), ]$mids # 0.009code 33も基本的にヒストグラムを解析することでどのデータを使うのが良いかを判断するための解析である。手を変え品を変え、色々とやってみるが結局わからなかった。
また、欠損値が全症例のうち9.9%以上10%以下のタンパク質を選んできて、その中でWDR59に着目し(欠損値が多ければ、すなわちmissForestにより代入された値が多ければどれでも良かった)、WDR59の発現量の分布もヒストグラムで見たりもした。でも、結局ここでもピンと来なかった。
# code 33
## Check distribution of imputed values.
### histogram of imputed value. the data was stored at list "hist_imputed_pre" and "hist_imputed_post".
hist_imputed_pre_argu <- hist(check_after_missForest$pre_argu,
breaks = 10000,
main = "Distribution of imputed values by missForest (pre normalization and back to arguments)",
xlab = "Expression level")
hist_imputed_pre <- hist(check_after_missForest$pre,
breaks = 10000,
main = "Distribution of imputed values by missForest (pre normalization)",
xlab = "Expression level")
hist_imputed_post <- hist(check_after_missForest$post,
breaks = 10000,
main = "Distribution of imputed values by missForest (post normalization)",
xlab = "Expression level")
### Distribution of the imputed NA values were stored at data frame "hist_imputed_pre_df" and "hist_imputed_post_df".
hist_imputed_pre_argu_df <- data.frame(mids = hist_imputed_pre_argu$mids, counts = hist_imputed_pre_argu$counts)
hist_imputed_pre_df <- data.frame(mids = hist_imputed_pre$mids, counts = hist_imputed_pre$counts)
hist_imputed_post_df <- data.frame(mids = hist_imputed_post$mids, counts = hist_imputed_post$counts)
### Take modal value for missForest results using the intensity before normalization.
hist_imputed_pre_argu_df[hist_imputed_pre_argu_df$counts == max(hist_imputed_pre_argu_df$counts), ]$counts # 3551
hist_imputed_pre_argu_df[hist_imputed_pre_argu_df$counts == max(hist_imputed_pre_argu_df$counts), ]$mids # -0.0075 # modal values when the break point is 10000
### Take modal value for missForest results using the intensity before normalization.
hist_imputed_pre_df[hist_imputed_pre_df$counts == max(hist_imputed_pre_df$counts), ]$counts # 1425
hist_imputed_pre_df[hist_imputed_pre_df$counts == max(hist_imputed_pre_df$counts), ]$mids # -0.005 # modal values when the break point is 10000
plot(hist_imputed_pre) # NA values for 9014 cases were imputed around 0 (modal value was 0.0175)
### Take modal value for missForest results using the normalized intensity.
hist_imputed_post_df[hist_imputed_post_df$counts == max(hist_imputed_post_df$counts), ]$counts # 1463
hist_imputed_post_df[hist_imputed_post_df$counts == max(hist_imputed_post_df$counts), ]$mids # 0.009 # modal values when the break point is 10000
plot(hist_imputed_post_df) # NA values for 1427 cases were imputed around 0 (modal value was 0.009)
## After prepare the data frame, clear unused memory as possible as it can be.
gc(T,T,T)
gc(T,T,T)
## Proteins contains the parcentage of NA around 0.1 were selected.
NA_more_than_0.1_gene <- merged_normalized_log2ratio_w_percent_na %>% filter(percent_na <= 0.1 & percent_na >= 0.099) %>% dplyr::select(percent_na)
## WDR59 was selected as a representative proteins for checking which dataset (missForest using before or after normalization) os better.
# result_missForest_pre_df_median_normalization_df["WDR59", ] %>% view()
## Checked with histogram. However, it is difficult to decide which dataset is better for missForest immputation.
### result_missForest_pre_argu_df_median_normalized_df
### result_missForest_pre_df_median_normalized_df
### result_missForest_post_df
hist(t(result_missForest_pre_argu_log2_df["WDR59", ]),
breaks = 1000,
main = "Distribution after missForest imputation then normalizaion again (pre normalization)",
xlab = "Expression level")
hist(t(result_missForest_pre_t_df["WDR59", ]),
breaks = 1000,
main = "Distribution after missForest imputation then normalizaion again (pre normalization)",
xlab = "Expression level")
hist(t(result_missForest_post_df["WDR59", ]),
breaks = 1000,
main = "Distribution after missForest imputation (post normalization)",
xlab = "Expression level")
## Other way to decide which dataset is better for missFortest imputation is comparison of difference between median values by CPTAC, missForest result using the dataset before normalization (data frame "result_missForest_pre_df_median_normalization_df"; the dataset of which is normalized again with median value from missForest result) and after normalization (data frame "result_missForest_post_df"; the dataset is median normalization by CPTAC or submitters).
### Check difference in the median value between data frame "result_missForest_pre_df_median_normalization_df" and "merged_mean_median_sd_3" (median value by CPTAC or submitter). The first column was used as the representative. but It was also difficult to decide which is better.
median(result_missForest_pre_argu_log2_df[,1]) # -1.055684
median(result_missForest_pre_t_df[,1]) # -1.075251 # median value of median-normalized data should be close to approximately 0. It looks OK.
median(result_missForest_post_df[,1]) # -1.073708 # I think it is weird. Why the value is close to median value by CPTAC or submitters?
median(merged_mean_median_sd_3[,1]) # -1.024735 # the value is median value that was calculated by CPTAC or submitters.
## The median value of first column of data frame "result_missForest_post_df" (-1.075444) is close to the median value by CPTAC or submitters (-1.024735)... I think it is weird. Why median value of median normalized intensity was close to median by CPTAC or submitters. The data frame "result_missForest_post_df" is MEDIAN NORMALIZED DATA and therefore the median value should be closed to 0 when the NA value means that the peptid was difficult to be detected and it resulted in the super low intensity.
## median_after_missForest <- data.frame(matrix(nrow = 1, ncol = ncol(result_missForest_post_df)))
## Next, calculate median value over the cases in both data frame "result_missForest_pre_df_median_normalization_df" and "result_missForest_post_df".
### required data;
#### matrix "result_missForest_pre_log2"
#### data frame "result_missForest_post_df"; the data frame is already normalized. it should be gotten back to the value before normalization, and then calculate median value.
#### data frame "merged_mean_median_sd_3"; this is mean, median, and sd for case of interest calculated by CPTAC or submitters.解析に採用したデータ
ここまで色々とデータを眺めてきて、今述べた通り「『CPTACで出している中央値に一番近いデータを得るには、正規化前の値を真数に戻した値、正規化前のlog2変換した値、正規化後のlog2変換(つまりCPTACの値そのまま)のどれを使えば良いのか?』を考えればいいじゃあないか?」ということに気がつく。
まず、missForestで得られた各データ(result_missForest_pre_argu_log2_df、result_missForest_pre_t_df、result_missForest_post_df)の列の中央値を計算し、その値をリストtempに入れる。そのリストtempの各要素には順にmedian_pre_argu、median_pre、median_postと名前を付けておく。そのリストをbind_rows()で横方向(列方向)に結合し、データフレームmedian_after_missForestを作成する。そして、さらにデータフレームmerged_mean_median_sd_3の中央値の部分(これはCPTACが計算した中央値)をデータフレームmedian_after_missForestにbind_rows()で結合する。そして、各データを使ってmissForestしたときの値の中央値(それぞれ列median_pre_argu、median_pre、median_post)とCPTACの中央値の差の絶対値をとり、その差が最小だった値が最も多いデータが、CPTACの中央値に一番近いimputationが出来たデータであると判断する。
その結果、正規化前の真数を使ってmissForestにより欠損値補完した場合に得られる中央値が、CPTACにより計算された中央値に一番近いという結果になった。これにより、この先で解析に使っていくデータはデータフレームresult_missForest_pre_argu_log2とした。これはcode 28で作成したデータフレームである。
# code 34
# temp <- list()
# temp[[1]] <- data.frame(t(apply(result_missForest_pre_df_median_normalization_df, 2, median))) # apply(MARGIN = 2) means apply the function (here, median) to each column. If MARGIN = 1, the function will be applied to each row.
# temp[[2]] <- data.frame(t(apply(result_missForest_post_df, 2, median))) # apply(MARGIN = 2) means apply the function (here, median) to each column. If MARGIN = 1, the function will be applied to each row.
# names(temp) <- c("median_pre", "median_post")
# result_missForest_post_mat <- as.matrix(result_missForest_post_df)
# view(result_missForest_post_mat[1:100, 1:100]) # Check data
# result_missForest_post_mat_2 <- sweep(x = result_missForest_post_mat, MARGIN = 2, STATS = merged_mean_median_sd_3_mat[2,], FUN = "+")
# view(result_missForest_post_mat_2[1:100, 1:100]) # Check data
temp <- list()
temp[[1]] <- data.frame(t(apply(result_missForest_pre_argu_log2_df, 2, median)))
temp[[2]] <- data.frame(t(apply(result_missForest_pre_t_df, 2, median)))
temp[[3]] <- data.frame(t(apply(result_missForest_post_df, 2, median)))
names(temp) <- c("median_pre_argu", "median_pre", "median_post")
## marge the columns.
median_after_missForest <- bind_rows(temp)
## name the columns.
rownames(median_after_missForest) <- names(temp)
colnames(median_after_missForest) <- colnames(result_missForest_post_df)
## Add median value by CPTAC or submitters to data frame "median_after_missForest".
median_after_missForest <- bind_rows(median_after_missForest, merged_mean_median_sd_3[2,]) %>% t() %>% data.frame()
## column "median_pre"; median value that is calculated with imputed NA value by missForest of intensity before normalization
## column "median_post"; median value that is calculated with imputed NA value by missForest of normalized intensity.
## column "Median"; median value that is calculated by CPTAC and submitters.
#
median_after_missForest$difference_pre_argu <- abs(median_after_missForest$median_pre_argu - median_after_missForest$Median)
median_after_missForest$difference_pre <- abs(median_after_missForest$median_pre - median_after_missForest$Median)
median_after_missForest$difference_post <- abs(median_after_missForest$median_post - median_after_missForest$Median)
median_after_missForest_difference <- colnames(median_after_missForest)[5:7]
median_after_missForest$smaller_difference_is <- apply(median_after_missForest[,5:7], 1, function(x) median_after_missForest_difference[which.min(x)])
table(median_after_missForest$smaller_difference_is)
## Following table is the benchmark of missForest imputation.
## missForest using arguments (inversed log2 value) before normalization got the median value that was more the closer to median value of CPTAC or submitters.
## difference_post difference_pre difference_pre_argu
## 1186 1612 2404
# median_after_missForest <- median_after_missForest %>% mutate(
# smaller_differece_is = case_when(
# difference_pre < difference_post ~ "pre",
# difference_pre > difference_post ~ "post",
# difference_pre == difference_post ~ "even",
# TRUE ~ NA)
# )
# table(median_after_missForest$smaller_differece_is)
# even post pre
# 2 218 4924
データの分布に有意差があるかどうか調べてみる
既にcode 34にて正規化前の真数のデータを使った中央値が、CPTACで計算された中央値に一番近いことがわかったので、このcode 35に意味はほとんど無いが、念のために各データでmissForestの前後の統計的分布に有意差があるか無いかをks.test()で、さらに、そのデータ全体の中央値にも差があるのか無いのかをwilcox.test()で確認しておく。本当ならば、ここで統計的分布に有意差が無いものを採用しようとしたが、結局、どのデータもmissForestの前後の統計分布に有意差があると出てしまった。こんなのでいいんだろうか….他はヒストグラムを出してみたり、データの中の最大値はどれか、なんかを見ている。
# code 35
hist(as.matrix(result_missForest_pre_argu_log2_df), breaks = 10000)
hist(as.matrix(result_missForest_pre_t_df), breaks = 10000)
hist(as.matrix(result_missForest_post_df), breaks = 10000)
## range of the imputed NA value
quantile(as.matrix(result_missForest_pre_argu_log2_df), probs = c(0.25, 0.5, 0.75)) # -0.5219597 -0.0705235 0.3350008
quantile(as.matrix(result_missForest_pre_t_df),, probs = c(0.25, 0.5, 0.75)) # -0.52889415 -0.07524554 0.33078758
quantile(as.matrix(result_missForest_post_df),, probs = c(0.25, 0.5, 0.75)) # -0.52895082 -0.07527012 0.33085971
## need wilcox.test and ks.test for comparison between before and after imputation.
ks.test(merged_analysis_before_normalization_ratio_mat_t, result_missForest_pre_argu$ximp) # p-value < 2.2e-16
wilcox.test(merged_analysis_before_normalization_ratio_mat_t, result_missForest_pre_argu$ximp) # p-value < 2.2e-16
ks.test(merged_log2ratio_6_mat_t, result_missForest_pre$ximp) # p-value < 2.2e-16
wilcox.test(merged_log2ratio_6_mat_t, result_missForest_pre$ximp) # p-value < 2.2e-16
ks.test(merged_normalized_log2ratio_6_mat_t, result_missForest_post$ximp) # p-value < 2.2e-16
wilcox.test(merged_normalized_log2ratio_6_mat_t, result_missForest_post$ximp) # p-value = 1.273e-07
#
hist(merged_analysis_before_normalization_ratio_mat_t, breaks = 100)
hist(result_missForest_pre_argu$ximp, breaks = 100)
hist(merged_analysis_before_normalization_ratio_mat_t[1:100, 1:100], breaks = 10000)
hist(result_missForest_pre_argu$ximp[1:100, 1:100], breaks = 10000)
max(merged_analysis_before_normalization_ratio_mat_t, na.rm = TRUE) # 327053101
max(result_missForest_pre_argu$ximp, na.rm = TRUE) # 327053101
#
hist(merged_log2ratio_6_mat_t, breaks = 100)
hist(result_missForest_pre$ximp, breaks = 100)
max(merged_log2ratio_6_mat_t, na.rm = TRUE) # 28.28495
max(result_missForest_pre$ximp, na.rm = TRUE) # 28.28495
#
hist(merged_normalized_log2ratio_6_mat_t, breaks = 100)
hist(result_missForest_post$ximp, breaks = 100)
max(merged_normalized_log2ratio_6_mat_t, na.rm = TRUE) # 28.95969
max(result_missForest_post$ximp, na.rm = TRUE) # 28.95969
# check maximum value.
max(merged_analysis_before_normalization_ratio_mat_t, na.rm = TRUE) # 327053101
range(merged_analysis_before_normalization_ratio_mat_t, na.rm = TRUE) # 3.510291e-09 3.270531e+08
max(merged_log2ratio_6_mat_t, na.rm = TRUE) # 28.28495
2^max(merged_log2ratio_6_mat_t, na.rm = TRUE) # 327053101code 34で、正規化前の真数をmissForestによる欠損値の補完に利用した場合、missForestの結果得られるデータの列の中央値は、CPTACが出している列の中央値に近いことがわかった。ここではそのデータを使える形に整える。code 28でマトリックスresult_missForest_pre_argu_log2を作成しているが、ここではそれを使って改めて正規化する。正規化にはCPTACにより算出された中央値を用いる。これはすでにlog2変換済のデータなので、CPTACにより算出された中央値をマトリックスresult_missForest_pre_argu_log2からsweep()を使って引き算する。そのようにしてマトリックスresult_missForest_pre_argu_df_median_normalizedを作成し、念のためにas.numeric()でそれらを数値にして、データフレームに変換することでデータフレームresult_missForest_pre_argu_df_median_normalized_dfを作成する。
マトリックスresult_missForest_pre_argu_df_median_normalizedの中央値は0.008310289だった。このデータはCPTACの中央値で正規化されているはずなので、CPTACが出している値に近ければ近いほどゼロに近づくはずである(中央値で正規化したということは、正規化後はその中央値はゼロになるはず)。データフレームに列名にファイル名、行名にタンパク質を付けて、write_tsv()で外部に保存しておく。列名と行名もcat()でテキストとして外部に保存しておく。
# code 36
## go to line 872 to check the matrix "result_missForest_pre_argu_log2".
## Normalization again. Later, check if the estimated NA value is close to median value by CPTAC or submitter.
## Remind; log2(merged_normalized_log2ratio_6_mat) - median = normalized_log2ratio
result_missForest_pre_argu_df_median_normalized <- sweep(x = result_missForest_pre_argu_log2,
MARGIN = 2,
STATS = merged_mean_median_sd_3_mat[2,],
FUN = "-")
# view(result_missForest_pre_argu_df_median_normalized[1:100, 1:100]) # check data
## Make sure the value of data frame "result_missForest_pre_df_median_normalization" is numeric.
# result_missForest_pre_argu_df_median_normalized <- apply(result_missForest_pre_argu_df_median_normalized, 2, as.numeric)
result_missForest_pre_argu_df_median_normalized <- apply(result_missForest_pre_argu_df_median_normalized, 2, as.numeric)
## Then the matrix "result_missForest_pre_df_median_normalization" transformed to data frame "result_missForest_pre_df_median_normalization_df"
# result_missForest_pre_argu_df_median_normalized_df <- data.frame(result_missForest_pre_argu_df_median_normalized)
result_missForest_pre_argu_df_median_normalized_df <- data.frame(result_missForest_pre_argu_df_median_normalized)
median(result_missForest_pre_argu_df_median_normalized) # 0.008310289, looks good because this dataset was centered with median, as the above.
## put column name (case) and row name(protein).
colnames(result_missForest_pre_argu_df_median_normalized_df) <- cases_pre_argu_normalization
rownames(result_missForest_pre_argu_df_median_normalized_df) <- proteins_pre_argu_normalization
## colnames(result_missForest_pre_argu_log2_df) <- cases_pre_argu_normalization
## rownames(result_missForest_pre_argu_log2_df) <- proteins_pre_argu_normalization
write_tsv(result_missForest_pre_argu_df_median_normalized_df, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/result_missForest_pre_argu_df_median_normalized_df.tsv")
cat(cases_pre_argu_normalization, file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/cases_pre_argu_normalization.txt", sep = "\n")
cat(proteins_pre_argu_normalization, file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/proteins_pre_argu_normalization.txt", sep = "\n")
ここまでのデータをRDataとして外部に保存しておく。
# code 37
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_3.RData")タンパク質発現プロファイルと臨床情報を結びつける
タンパク質発現のデータを整えたら、次は臨床情報の整理である。問題は一つのタンパク質発現プロファイル(一つの列)に対して複数の症例が割当られていたり、ある症例では正常組織と腫瘍組織の両方を一つのサンプル(プールされたサンプル)で解析されていたりすることである。正常組織と腫瘍組織のプールって、一体どういうことやねん。そんなのどう考えたっておかしいやんか。症例数が少ないことに加えて、こういうところがCPTACはTCGAよりに比べて明らかに未発達だなと感じるところである。TCGAもそうだが、CPTACの解析はこのタンパク質発現プロファイルと臨床情報を結びつける作業に多くの時間を費やすはずである。
ここでやっていることは、口で言うのは非常に簡単である。まずはbiospeciemen(biospecimen_manifestとついているtsvファイル)から、症例のタンパク質発現プロファイルではないもの(例えばCell lineやxenograftなど)や、それさえもよくわからない謎のタンパク質発現プロファイルを除き、区別できるタンパク質発現プロファイルの列名とbiospeciemenの列Aliquot Submitter IDを結合させる。続いて、それらとclinical(clinical_manifestとついているtsvファイルもしくはcsvファイル)とfollowup(followup_manifestとついているtsvファイルもしくはcsvファイル)を列Case IDで結合する。「はじめに」の章でも書いたが、CPTACにはタンパク質発現プロファイルとそのファイルとリンクする臨床情報を結びつけることができない症例が結構ある。これらを見分けるためにもstudy(study_manifestとあるtsvファイルもしくはcsvファイル)を見ながら、重複している情報をまとめていく必要がある。このbiospeiemen、clinical、followupの整理は随分と手間である。
この解析では、treatment(treatment_manifestとあるtsvファイルもしくはcsvファイル)とexposure(exposure_manifestとあるtsvファイルもしくはcsvファイル)は使用しなかった。上記clinical、followupのファイルと結合しようとすると、少しややこしいことになる。その理由は、一人の患者でいくつもの抗癌剤治療など、複数の治療を行っているためである。こればかりは情報が重複するのは当然であり、この解析ではシンプルにタンパク質発現量と疾患ごとの生存期間が知りたいので、それらは解析しなかった。もちろん、これらに興味があれば、別途データを整理して解析に用いる必要がある。
まずは予めダウンロードしたそれぞれのマニフェストファイルを読み込んで、データフレームbiospeciemen、clinical、followup、treatment、exposure、study、manifestを作成する。
# code 38
# Read clinical information.
## biospeciemen
biospeciemen <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_biospecimen_manifest_10202024_102342.tsv")
nrow(biospeciemen) # 5228 cases
## clinical
clinical <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_clinical_manifest_10202024_102355.tsv")
nrow(clinical) # 3563 cases
## followup
followup <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_clinical_followup_manifest_10202024_102355.tsv")
nrow(followup) # 279 cases
## treatment
treatment <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_clinical_treatment_manifest_10202024_102355.tsv")
nrow(treatment) # 84 cases
## exposure
exposure <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_clinical_exposure_manifest_10202024_102355.tsv")
nrow(exposure) # 1222 cases
## study
study <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_study_manifest_10202024_102331.tsv")
nrow(study) # 55 studies
## manifest file for download protein expression data
manifest <- read_tsv("/mnt/seqdata/public_data/Blog/CPTAC/PDC_file_manifest_10202024_102427.tsv")
nrow(manifest) # 278 studies
code 39では、各データフレームの列名から不要な文字(特にスペース)をアンダーバーなどの問題の少ない文字に置き換えたり、今後見やすく列を並び替えたり、各データフレームの列Case IDを”Case_ID”に文字列に統一したりする。
ここで、データフレームbiospeciemenの列Aliquot_Submitter_IDに入っている文字列のうち、ハイフンをアンダーバーに置き換えて、その値を新しく列Aliquot_Submitter_ID_2とする。このAliquot_Submitter_IDは、各症例もしくは各サンプルのタンパク質発現プロファイルの列名にも使用されている。つまり、データフレームbiospeciemenの列Aliquot_Submitter_IDとここで作成する列Aliquot_Submitter_ID_2は、データフレームbiospeciemenとタンパク質発現プロファイルのデータフレームを結合するために必要になる。
このcode 39の最後に、full_join()で各データフレームを結合し、データフレームclinical_info_mergedを作成するが、これは全体を一気に見るためのもので、後々の解析には使用しない。どの列にどんな値が入っているのかを確認したり、タンパク質発現プロファイル名を取得するために使用する。
## code 39
### Remove unnecessary characters from the columns of clinical information.
print("------------------------------biospeciemen----------------------------------------------")
colnames(biospeciemen)[1:10]
print("------------------------------treatment----------------------------------------------")
colnames(treatment)[1:10]
print("------------------------------exposure----------------------------------------------")
colnames(exposure)[1:10]
print("------------------------------followup----------------------------------------------")
colnames(followup)[1:10]
print("------------------------------clinical----------------------------------------------")
colnames(clinical)[1:10]
### biospeciemen
### "biospeciemen_renamed" will be used for matching the file name of protein expression later.
colname_biospeciemen <- colnames(biospeciemen)
colname_biospeciemen <- gsub(pattern = " ", "_", colname_biospeciemen)
colnames(biospeciemen) <- colname_biospeciemen
biospeciemen$Aliquot_Submitter_ID_2 <- gsub(pattern = "-", replacement = "_", x = biospeciemen$Aliquot_Submitter_ID)
biospeciemen <- biospeciemen %>% select(c(1,2, ncol(biospeciemen), 3:(ncol(biospeciemen)-1)))
ncol(biospeciemen)
### treatment
colname_treatment <- colnames(treatment)
colname_treatment <- gsub(pattern = " ", "_", colname_treatment)
colnames(treatment) <- colname_treatment
### exposure
colname_exposure <- colnames(exposure)
colname_exposure <- gsub(pattern = " ", "_", colname_exposure)
colnames(exposure) <- colname_exposure
### followup
colname_followup <- colnames(followup)
colname_followup <- gsub(pattern = " ", "_", colname_followup)
colnames(followup) <- colname_followup
### rename column "Case_Id", "Case_Submitter_Id", "Follow_Up_Submitter_Id", "Follow_Up_Id" for merge with other data frame.
followup <- followup %>% dplyr::rename("Case_ID" = "Case_Id")
followup <- followup %>% dplyr::rename("Case_Submitter_ID" = "Case_Submitter_Id")
followup <- followup %>% dplyr::rename("Follow_Up_Submitter_ID" = "Follow_Up_Submitter_Id")
followup <- followup %>% dplyr::rename("Follow_Up_ID" = "Follow_Up_Id")
### clinical
colname_clinical <- colnames(clinical)
colname_clinical <- gsub(pattern = " ", "_", colname_clinical)
colnames(clinical) <- colname_clinical
## study
colname_study <- colnames(study)
colname_study <- gsub(pattern = " ", "_", colname_study)
colnames(study) <- colname_study
study <- study %>% dplyr::rename("Case_number" = "Cases_#") # rename column "Case_Id" to "Case_ID" for merge with other data frame.
clinical_info_list <- list(treatment, exposure, followup, clinical)
names(clinical_info_list) <- c("treatment", "exposure", "followup", "clinical")
# Retrieve column name again. Becuase the "_" was removed from the columns at the above.
colname_biospeciemen_df <- data.frame(column = colnames(biospeciemen))
colname_clinical_df <- data.frame(column = colnames(clinical))
colname_treatment_df <- data.frame(column = colnames(treatment))
colname_exposure_df <- data.frame(column = colnames(exposure))
colname_followup_df <- data.frame(column = colnames(followup))
colname_study_df <- data.frame(column = colnames(study))
# this data frame will be used for checking the data.
clinical_info_merged <- full_join(biospeciemen, clinical, by = "Case_ID", suffix = c("", "_clinical"))
nrow(clinical_info_merged) # 5306
clinical_info_merged <- full_join(clinical_info_merged, followup, by = "Case_ID", suffix = c("", "_followup"))
nrow(clinical_info_merged) # 5306
clinical_info_merged <- full_join(clinical_info_merged, treatment, by = "Case_ID", suffix = c("", "_treatment"))
nrow(clinical_info_merged) # 5339
clinical_info_merged <- full_join(clinical_info_merged, exposure, by = "Case_ID", suffix = c("", "_exposure"))
nrow(clinical_info_merged) # 5339code 40では、ひとまず解析に使用できなさそうな症例(ゼノグラフトなどがあるので、正しくは症例ではなく、scpeicemen;試料とかサンプルという表現が当てはまると思う。今後、こういった謎症例は「試料」や「サンプル」と呼ぶ)を除く。最初に、データフレームbiospeciemenの列Tissue_Typeが「Not Reported」になっている症例は、一体何者なのか確認する。そのために列Tissue_Typeが「Not Reported」の症例をデータフレームbiospeciemenから抽出して、新しくデータフレームcheck_Tissue_Type_not_reportedを作成し、その中身をtable()で逐一確認していく。そうすると列Sample_Typeに「Cell Lines」とか「Xenograft Tissue」とか、症例ではないものがあるとわかる。これらは後ほど除くとして、ここでは放っておく。また、その中にさらに「Not Reported」というサンプルがあるようだ。そこでデータフレームcheck_Tissue_Type_not_reportedから列Sample_Typeが「Not Reported」のサンプルを抽出してきて、データフレームcheck_Tissue_Type_Sample_Type_not_reportedを作成し、table()で確認する。しかし、結局のところ列Sample_Typeが「Not Reported」のサンプルは列Tissue_Typeも「Not Reported」
で、一体何なのかは同定出来なかった。そうなってくると、これを個別に抽出し、Case_IDなど他の情報も見てみる必要がある。なので、データフレームclinical_info_mergedの列Case_IDとデータフレームcheck_Tissue_Type_Sample_Type_not_reportedの列Case_IDを%in%で同一の症例を抽出してきて、データフレームcheck_Tissue_Type_Sample_Type_not_reported_2を作成し、それらが必要かどうかを全体的な情報から判断する。その結果わかったのが、これらは多くがQC用のサンプルだったり、細胞株だったり、何かしらのコントロールやリファレンス(おそらく質量分析用)だったり、挙句の果にはWithdrawnだった事である。しかし、中には症例らしいものも含まれているので、なんとかしてそれらを入れた方が良い様に思う。そこで、データフレームcheck_Tissue_Type_Sample_Type_not_reported_2から、列Vital_Status、列Days_to_Last_Follow_Up、列Days_to_Deathが「NA」ではないサンプル、つまり、生存しているときにフォローアップされている症例の可能性があるサンプルを抽出してきてデータフレームcheck_Tissue_Type_Sample_Type_not_reported_3を作成し、さらにそこから、データを見やすくするために列Case_ID、列Case_Submitter_ID、列Vital_Status、列Days_to_Last_Follow_Up、列Days_to_Deathを選んできてデータフレームcheck_Tissue_Type_Sample_Type_not_reported_4を作成し、それらを見ながら、これらのサンプルは生存期間解析に使える症例なのか、そうではないのかを判断する。その結果、どうやら列Case_IDの値が「9955c013-c8fd-4315-8946-f0c41edb024d」と「10784784-5101-4f5e-aefa-1311e0000fdf」は解析に使用できそうだった。今後、この症例(もしくは何かのサンプル)はどこかで除去される可能性もあるが、これらはひとまず方っておいて、後ほど別途整形する。
生存期間解析に使用できそうな症例が大体把握できたので、次はデータフレームbiospeciemenから実際に余計なサンプルを除いていく。まず、データフレームbiospeciemenの列Case_Statusと列Sample_Statusの両方が「Disqualified」の症例を除き、データフレームbiospeciemen_1を作成する。
これにしたって、予め除いてから登録しろよって感じである。少なくともタンパク質発現プロファイルの解析を行うためには症例とかサンプルの状態が悪いという意味だろうと思う。なんでこれを残しているんだろうか…CPTACはこういうところが未発達だと思うところである。つまり、汎用的ではないデータをわざわざ登録しているマヌケな研究機関や研究者が居るってことである。これはもう個人的にはデータのコンタミネーションである。だって、臨床情報としてDisqualifiedだったら、どんな解析にだって使えないはずである。言うてても仕方がない。
データフレームbiospeciemenから不要なサンプルを除いていき、データフレームbiospeciemen_4を作成する。
| 新しいデータフレーム | 元のデータフレーム | 列 | 値 | |
|---|---|---|---|---|
| biospeciemen_1 | biospeciemen | Case_Status Sample_Status | Disqualified | 除去 |
| biospeciemen_2 | biospeciemen_1 | Tissue_Type | Not Reported | 除去 |
| biospeciemen_3 | biospeciemen_2 | Sample_Type | Cell Lines Xenograft Tissue | 除去 |
| biospeciemen_4 | biospeciemen_3 | Sample_Type | Normal Adjacent Tissue Primary Tumor Solid Tissue Normal Tumor | 残す |
また、データフレームbiospeciemenから、列Case_IDの値が「9955c013-c8fd-4315-8946-f0c41edb024d」と「10784784-5101-4f5e-aefa-1311e0000fdf」を抽出し、それらをデータフレームbiospeciemen_5とし、それをデータフレームbiospeciemen_4とbind_rows()で縦方向(行方向)に結合し、データフレームbiospeciemen_6を作成する。
# code 40
# Important;
## biospeciemen dataframe contains "Tissue_Type" column, which has "Normal" or "Tumor".
table(biospeciemen$Tissue_Type)
# Normal Not Reported Tumor
# 1014 1314 2900
# check what is the observation with "Not Reported" in "Tissue_Type" column.
check_Tissue_Type_not_reported <- biospeciemen %>% filter(Tissue_Type == "Not Reported")
table(biospeciemen$Tissue_Type)
table(biospeciemen$Sample_Type)
table(biospeciemen$Tissue_Type, biospeciemen$Sample_Type)
table(check_Tissue_Type_not_reported$Sample_Type)
# Cell Lines Normal Adjacent Tissue Not Reported Primary Tumor Solid Tissue Normal
# 119 3 76 905 169
# Xenograft Tissue
# 42
# At least, "Cell Lines" and "Xenograft Tissue" values in "Sample_Type" column are not required. On the other hand, the obsercation with "Not Reported" in "Sample_Type" columns is unknown. Therefore check them.
check_Tissue_Type_Sample_Type_not_reported <- check_Tissue_Type_not_reported %>% filter(Sample_Type == "Not Reported")
table(check_Tissue_Type_Sample_Type_not_reported$Sample_Type)
# Not Reported
# 76
table(check_Tissue_Type_Sample_Type_not_reported$Tissue_Type)
# Not Reported
# 76
# I don't know what is the observation with "Not Reported" value in Sample_Type" columns. Extract them from "clinical_info_merged" data frame.
check_Tissue_Type_Sample_Type_not_reported_2 <- clinical_info_merged[clinical_info_merged$Case_ID %in% check_Tissue_Type_Sample_Type_not_reported$Case_ID, ]
check_Tissue_Type_Sample_Type_not_reported_3 <- check_Tissue_Type_Sample_Type_not_reported_2 %>% filter(
Vital_Status != "NA"|
Days_to_Last_Follow_Up != "NA" |
Days_to_Death != "NA")
check_Case_Submitter_ID <- check_Tissue_Type_Sample_Type_not_reported_3 %>% dplyr::select(c(
Case_Submitter_ID,
Case_Submitter_ID_clinical,
Case_Submitter_ID_followup,
Case_Submitter_ID_treatment,
Case_Submitter_ID_exposure))
check_Tissue_Type_Sample_Type_not_reported_4 <- check_Tissue_Type_Sample_Type_not_reported_3 %>% select(
Case_ID,
Case_Submitter_ID,
Vital_Status,
Days_to_Last_Follow_Up,
Days_to_Death)
# Of the observation with "Not Reported" value in Sample_Type" columns, "Case_ID" == "9955c013-c8fd-4315-8946-f0c41edb024d" and "10784784-5101-4f5e-aefa-1311e0000fdf" can be used for survival analysis, because their "Vital Status" value is "Dead" and they have the values in "Days to Last Follow Up" and "Days to Death" column. Check the values in `Vital Status`, `Days to Last Follow Up`, `Days to Death`, `Primary Diagnosis`, `Site of Resection or Biopsy`, `Tissue or Organ of Origin`, `Primary Site`, `Disease Type` columns in "clinical_info_merged" data frame of these two cases.
table(clinical_info_merged$Primary_Diagnosis) %>% data.frame() %>% view()
table(biospeciemen$Case_Status)
table(biospeciemen$Sample_Status)
biospeciemen_1 <- biospeciemen %>% filter(Case_Status != "Disqualified" & Sample_Status != "Disqualified")
##
biospeciemen_2 <- biospeciemen_1 %>% filter(Tissue_Type != "Not Reported")
table(biospeciemen_2$Tissue_Type, biospeciemen_2$Sample_Type)
## I dont know why but OR (|) does not work well in the following code. Therefore I run filter function twice.
biospeciemen_3 <- biospeciemen_2 %>% filter(Sample_Type != "Cell Lines")
biospeciemen_3 <- biospeciemen_3 %>% filter(Sample_Type != "Xenograft Tissue")
table(biospeciemen_3$Tissue_Type, biospeciemen_3$Sample_Type)
table(biospeciemen_3$Sample_Type) %>% data.frame() %>% view()
# # Following values in "Sample_Type" column will be required
# "Normal Adjacent Tissue"
# "Primary Tumor"
# "Solid Tissue Normal"
# "Tumor"
biospeciemen_4 <- biospeciemen_3 %>% filter(Sample_Type == "Normal Adjacent Tissue" |
Sample_Type == "Primary Tumor" |
Sample_Type == "Solid Tissue Normal" |
Sample_Type == "Tumor")
table(biospeciemen_4$Sample_Type)
## See line 1099, as describing below.
## Of the observation with "Not Reported" value in Sample_Type" columns, "Case_ID" == "9955c013-c8fd-4315-8946-f0c41edb024d" and "10784784-5101-4f5e-aefa-1311e0000fdf" can be used for survival analysis, because their "Vital Status" value is "Dead" and they have the values in "Days to Last Follow Up" and "Days to Death" column. Check the values in `Vital Status`, `Days to Last Follow Up`, `Days to Death`, `Primary Diagnosis`, `Site of Resection or Biopsy`, `Tissue or Organ of Origin`, `Primary Site`, `Disease Type` columns in "clinical_info_merged" data frame of these two cases.
biospeciemen_5 <- biospeciemen %>% filter(Case_ID == "9955c013-c8fd-4315-8946-f0c41edb024d" | Case_ID == "10784784-5101-4f5e-aefa-1311e0000fdf")
# table(biospeciemen_renamed_5$Sample_Type)
#
check_biospeciemen_5 <- clinical_info_merged %>% filter(Case_ID == "9955c013-c8fd-4315-8946-f0c41edb024d" | Case_ID == "10784784-5101-4f5e-aefa-1311e0000fdf")
check_biospeciemen_5_2 <- check_biospeciemen_5 %>% select(
Case_ID, Case_Submitter_ID, Vital_Status, Days_to_Last_Follow_Up, Days_to_Death, Primary_Diagnosis, Site_of_Resection_or_Biopsy, Tissue_or_Organ_of_Origin, Primary_Site, Disease_Type)
biospeciemen_6 <- bind_rows(biospeciemen_4, biospeciemen_5)
# the cases recorded in "biospeciemen_renamed_6" data frame will be analyzed for further analysis, especially violin plot for expression analysis
# Again, "Case_ID" == "9955c013-c8fd-4315-8946-f0c41edb024d" and "10784784-5101-4f5e-aefa-1311e0000fdf" in "biospeciemen_renamed_6" data frame is pancreatic ductal carcinoma.次に、タンパク質発現プロファイル名を整理して、code 40で作成したサンプルの情報と結合出来るようにする。ここではデータフレームresult_missForest_pre_argu_df_median_normalized_dfから列名だけをcolnames()で取得し、データフレームfile_name_case_idとする。そしてこのデータフレームに列IDとして、1から順番に番号を振る。それらはもしかしたらどこかで使うかもしれないので、write_tsv()で書き出しておく。
# code 41
# NEED missForest RESULT HERE!!!!
# This is file name of analysis dataset.
# The column name contains "Aliquot Submitter ID" at biospeciemen_manifest file.
file_name_case_id <- colnames(result_missForest_pre_argu_df_median_normalized_df) %>% data.frame()
colnames(file_name_case_id) <- "name"
file_name_case_id$ID <- seq(1:nrow(file_name_case_id))
# write_tsv(sample, "E:/Data B049 0010 19/sample_name.tsv") # Use this tsv file to check it.
write_tsv(file_name_case_id, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/file_name_case_id.tsv") # Use this tsv file to check it.データフレームfile_name_case_idの列nameには、タンパク質発現プロファイル名が入っている。この値は、元のタンパク質発現プロファイル名に、例えばPDC000110というようなPDCから始まる番号(PDC study ID)を繋いだ文字列である。PDC study IDと直後のアンダーバー以降の文字列は、biospeciemenのAliquot Submitter ID(この解析ではcode 39で作成した列Aliquot_Submitter_ID_2の値)と一致するので、それをfile_name_case_idの列name(タンパク質発現プロファイル名)から抽出する。ここでも正規表現を使って文字列を操作するわけだが、これは全部chatGPTに聞いたらよい。まず、”^[^_]+_”は、”最初のアンダーバー以降の文字列”、”_Log_Ratio$”は”文字列の最後が_Log_Ratio”、”_.*”は”アンダーバー以降全部”という意味である。なんか、よくわからん。こんなもんchatGPTに教えてもらおう。これらを使って、データフレームfile_name_case_idに、列possible_Aliquot_Submitter_IDを作成し、そこに、例えば「9955c013_c8fd_4315_8946_f0c41edb024d」といったような値を、そして、列PDC_Study_IDに、例えば「PDC000110」というような値を入れる。
最後に、table()でデータフレームfile_name_case_idの列possible_Aliquot_Submitter_IDと、データフレームbiospeciemenもしくはデータフレームbiospeciemen_6の列Aliquot_Submitter_ID_2で、どのくらい一致、不一致の例があるのか確認しておく。そうすると、一致しないものも随分多いようだ。
# code 42
file_name_case_id$possible_Aliquot_Submitter_ID <- gsub(pattern = "^[^_]+_", replacement = "", x = file_name_case_id$name)
file_name_case_id$possible_Aliquot_Submitter_ID <- gsub(pattern = "_Log_Ratio$", replacement = "", x = file_name_case_id$possible_Aliquot_Submitter_ID)
file_name_case_id$PDC_Study_ID <- gsub(pattern = "_.*", replacement = "", x = file_name_case_id$name)
table(file_name_case_id$possible_Aliquot_Submitter_ID %in% biospeciemen$Aliquot_Submitter_ID_2)
# FALSE TRUE
# 2169 3033
table(file_name_case_id$possible_Aliquot_Submitter_ID %in% biospeciemen_6$Aliquot_Submitter_ID_2)
# FALSE TRUE
# 3111 2091 タンパク質発現プロファイル名を保持したデータフレームfile_name_case_idの列possible_Aliquot_Submitter_IDと、解析に使えなさそうな症例を除去したデータフレームbiospeciemen_6の列Aliquot_Submitter_ID_2(Aliquot_Submitter_IDのハイフンをアンダーバーに直したもの)が一致しているタンパク質発現プロファイル名を選んできて、それをデータフレームfile_name_case_id_2とする。列Aliquot_Submitter_IDの他に重要な列は、列Case_IDである。列Case_IDは、臨床情報の各データフレームclinical(診断された情報や患者の情報)やfollowup(フォローアップの期間)を互いに結合するために使用できる。一番面倒なのは、タンパク質発現プロファイル名(すなわち列Aliquot_Submitter_ID、列Aliquot_Submitter_ID_2、タンパク質発現プロファイル名から自分で作成した列possible_Aliquot_Submitter_ID)と、この列Case_IDを結びつけることが出来ないことと思う。タンパク質発現プロファイルの列名と一発で結合出来るようなフォーマットに統一してほしいところである。どの様に結合するかといえば、この解析で行っているように各タンパク質発現プロファイルの列名データフレームbiospeciemenの列Aliquot_Submitter_IDを結合し、それに対応する列Case_IDを得てから、それを使ってデータフレームclinicalとデータフレームfollowupを結合する、という感じである。データフレームfile_name_case_id_2は、今後の解析に使用できそうなタンパク質発現プロファイルの列名が入っている。それに対応するであろう列Case_IDの値をなんとかして入れる必要がある。だからといって、inner_join()とかでは、一つ一つの値を選べるわけではないので、上手く結合することが出来ない。なので、リストfile_name_case_id_2_listの各要素に対して一つの列Aliquot_Submitter_IDの値を持つ例を入れていって(おそらく各要素に複数の値が入る可能性がある)、それに一致する列Case_IDを取得し、最後にbind_rows()でリストを縦方向に結合させ、データフレームfile_name_case_id_2_dfを作成する。これで、各タンパク質発現プロファイルの列名に対応する列Case_IDの値を入れることが出来る。このようにしても、おかしなタンパク質発現プロファイルもしくはサンプルが見つかる。どうやら、データフレームfile_name_case_id_2_dfの列possible_Aliquot_Submitter_IDの値が「618」と「CPT0162600005」で、区別できないタンパク質発現プロファイルがあるらしい。この2つの症例に関しては、ここ(CPTACのウェブページ;https://pdc.cancer.gov/pdc/)の以下の写真のところに、列Aliquot_Submitter_IDもしくは列Aliquot_Submitter_ID_2の値が「618」もしくは「CPT0162600005」の症例の列Case_IDの値を入力して、個別にどんな症例でどんなサンプルなのかを確認する必要がある。その結果、どうやら「CPT0162600005」は列Case_IDが単に重複している、つまり、同一患者由来であることがわかり、一方、「618」は複数のプロジェクトで同じような列名のタンパク質発現プロファイルがあることがわかった。この列Aliquot_Submitter_ID(または列Aliquot_Submitter_ID_2)が「618」の症例が問題である。こういう症例をinner_join()やfull_join()で結合してしまうと、最終的に別の症例の情報が結合される可能性があるので、要注意である。
この「618」に関しては、データフレームfile_name_case_id_2_dfを見るのが良いのではないかと思う。データフレームfile_name_case_id_2_dfをview()などで表示して、「618」で検索してみると、その文字列を持つ症例の情報が色々みることがわかる。その中でも、列PDC_study_IDと列Case_IDが有用であり、その値を元に上記のCPTAC(というかPDC; Proteomics Data Portal)のページで検索してみると良い。この列Aliquot_Submitter_IDもしくは列Aliquot_Submitter_ID_2の値が「618」の症例は、PDC_study_IDが「PDC000446」であるべきらしい。そしてこの例は、データフレームfile_name_case_id_2_df上では列Case_IDが「7b818a1e-39c0-464f-8f5c-8ffdd14217b7」と「7a598673-1168-11ea-9bfa-0a42f3c845fe」が関連付けてある。
データフレームfile_name_case_id_2_dfを618で検索したところ。
これを上記のCPTAC(というかPDC; Proteomics Data Portal)のページで検索すると、どうやら「7a598673-1168-11ea-9bfa-0a42f3c845fe」はPDC Study IDがPDC000198であり、PDC000446ではないことがわかった(以下の画像)。
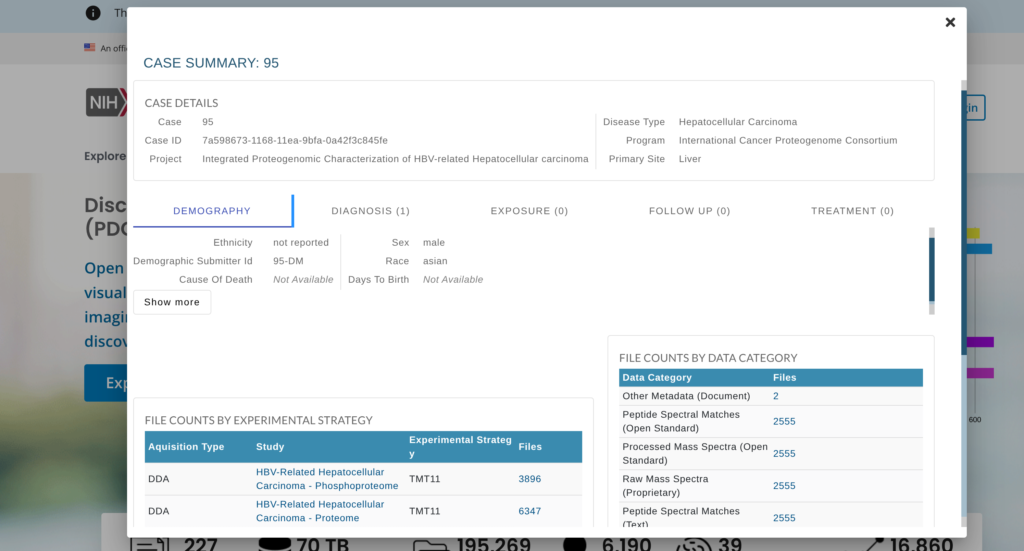
FILE COUNTS BY EXPERIMENTAL STRATEGYのHBV-Related Hepatocellular Carcinoma – Proteomeをクリックする。

つまり、この列Aliquot_Submitter_ID(または列Aliquot_Submitter_ID_2)が「618」の症例は、列Case_IDの値が「7b818a1e-39c0-464f-8f5c-8ffdd14217b7」が正解ということになる。これらの情報をもとに、データフレームfile_name_case_id_2_dfから列Case_ID、列PDC_Study_ID、列Aliquot_Submitter_ID_2の値がこの2つの症例に一致するものを抽出してきて、データフレームfile_name_case_id_2_df_duplicatedを作成し(データフレーム名が紛らわしい。実際はduplicatedを解消したデータフレームである)し、同時に、上記のような確認なしで同定できた症例のみで構成されたデータフレームfile_name_case_id_2_df_uniqueを作成し、それら2つをbind_rows()で縦方向に結合させて、新しくデータフレームfile_name_case_id_3を作成した。最後にこのデータフレームfile_name_case_id_3で無駄に重複している症例が無いことを確認しておいた。
# code 43
# 2026 04 18
file_name_case_id_2 <- file_name_case_id[file_name_case_id$possible_Aliquot_Submitter_ID %in% biospeciemen_6$Aliquot_Submitter_ID_2 == TRUE,]
table(file_name_case_id_2$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq == 1) %>% nrow() # 2091 cases
table(file_name_case_id_2$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0 cases
#
file_name_case_id_2_list <- list()
#
for(i in 1:nrow(biospeciemen_6)){
#
file_name_case_id_2_list[[i]] <- file_name_case_id_2[file_name_case_id_2$possible_Aliquot_Submitter_ID %in% biospeciemen_6$Aliquot_Submitter_ID_2[i] == TRUE,]
#
file_name_case_id_2_list[[i]] <- file_name_case_id_2_list[[i]] %>% mutate(
ID = file_name_case_id_2_list[[i]]$ID,
Case_ID = biospeciemen_6$Case_ID[i],
Aliquot_Submitter_ID_2 = biospeciemen_6$Aliquot_Submitter_ID_2[i])}
#
names(file_name_case_id_2_list) <- biospeciemen_6$Aliquot_Submitter_ID_2
file_name_case_id_2_df <- bind_rows(file_name_case_id_2_list)
table(file_name_case_id_2_df$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq == 1) %>% nrow() # 2089 cases
table(file_name_case_id_2_df$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 2 cases
table(file_name_case_id_2_df$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% view()
# 618
# CPT0162600005
table(file_name_case_id_2_df$name) %>% data.frame() %>% filter(Freq > 1) %>% view()
biospeciemen_6[str_detect(string = biospeciemen_6$Aliquot_Submitter_ID_2,
pattern = "618"),] %>% view()
# The overlapped cases CPT0162600005 has same Case_ID. This duplication can be just removed.
# check at here manually; https://proteomic.datacommons.cancer.gov/pdc/
# Aliquot_Submitter_ID_2 == CPT0162600005
# PDC_Study_ID == PDC000270
# Case_ID == 10784784-5101-4f5e-aefa-1311e0000fdf
biospeciemen_6[str_detect(string = biospeciemen_6$Aliquot_Submitter_ID_2,
pattern = "CPT0162600005"),] %>% view()
# In "file_name_case_id_2", "possible_Aliquot_Submitter_ID" should be same as "Aliquot_Submitter_ID_2"
# file_name_case_id_3$Aliquot_Submitter_ID_2 <- file_name_case_id_2$possible_Aliquot_Submitter_ID
# file_name_case_id_3 <- file_name_case_id_2 %>% mutate(
# Aliquot_Submitter_ID_2 = file_name_case_id_2$possible_Aliquot_Submitter_ID)
file_name_case_id_2_df_unique <- file_name_case_id_2_df[
(file_name_case_id_2_df$possible_Aliquot_Submitter_ID != "618" & file_name_case_id_2_df$possible_Aliquot_Submitter_ID != "CPT0162600005"),]
nrow(file_name_case_id_2_df_unique) # 2089 cases
file_name_case_id_2_df_duplicated <- file_name_case_id_2_df[
(file_name_case_id_2_df$Case_ID == "7b818a1e-39c0-464f-8f5c-8ffdd14217b7" & file_name_case_id_2_df$PDC_Study_ID == "PDC000446" & file_name_case_id_2_df$Aliquot_Submitter_ID_2 == "618") |
(file_name_case_id_2_df$Case_ID == "10784784-5101-4f5e-aefa-1311e0000fdf" & file_name_case_id_2_df$PDC_Study_ID == "PDC000270"& file_name_case_id_2_df$Aliquot_Submitter_ID_2 == "CPT0162600005"),] %>% distinct(Aliquot_Submitter_ID_2, .keep_all = TRUE)
nrow(file_name_case_id_2_df_duplicated) # 2 cases
file_name_case_id_3 <- bind_rows(file_name_case_id_2_df_unique, file_name_case_id_2_df_duplicated)
nrow(file_name_case_id_3) # 2091 cases
table(file_name_case_id_3$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq == 1) %>% nrow() # 2091 cases
table(file_name_case_id_3$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0 casescode 44では、データフレームfile_name_case_idの列possible_Aliquot_Submitter_IDの値が、データフレームbiospeciemen_6の列Aliquot_Submitter_ID_2の値と一致しないものについて見る。それらをデータフレームfile_name_case_id_2_FALSEとして作成した。このデータフレームfile_name_case_id_2_FALSEと、データフレームstudy(これはcode 38で作成した)とinner_join()する。列PDC_Study_IDを軸にして結合されるはずである。
# code 44
#
file_name_case_id_2_FALSE <- file_name_case_id[file_name_case_id$possible_Aliquot_Submitter_ID %in% biospeciemen_6$Aliquot_Submitter_ID_2 == FALSE,]
#
check_file_name_Aliquot_Submitter_ID_inconsistency <- inner_join(file_name_case_id_2_FALSE, study)
# See following data frame
biospeciemen # original of biospeciemen
biospeciemen_6 # data with values for survival analysis
file_name_case_id # file names of the missForest results.
file_name_case_id_2 # Of the file name of the missForest results, the cases that can be used for survival analysis (with values for survival analysis).
file_name_case_id_2_FALSE # Of the file name of the missForest results, the cases that can NOT be used for survival analysis (WITHOUT values for survival analysis).
# add column "biospeciemen_6$Aliquot_Submitter_ID_2" into file_name_case_id_2
temp_biospeciemen_6_Aliquot_Submitter_ID_2 <- biospeciemen_6 %>% select(c("Aliquot_Submitter_ID_2"))
nrow(temp_biospeciemen_6_Aliquot_Submitter_ID_2) # 3301 casesこのデータフレームfile_name_case_id_2_FALSE(データフレームbiospeciemen_6の列Aliquot_Submitter_IDや列Aliquot_Submitter_ID_2とタンパク質発現プロファイル名が一致しなかったもの)から出来る限り解析できそうな症例を抽出したい。code 45では、データフレームfile_name_case_id_2_FALSEの列possible_Aliquot_Submitter_IDの値(文字列)に、データフレームbiospeciemen_6の列Aliquot_Submitter_ID_2の値(文字列)が含まれていた場合(だからここではstr_detect()を使う。code 43では一致するものがほしかったので%in%を使った)にリストfile_name_case_id_4_listの各要素のそのデータフレームを入れてき、そして、データフレームbiospeciemen_6の一致する列Case_IDと列Aliquot_Submitter_ID_2の値を加えていき、最後にbind_rows()でリストfile_name_case_id_4_listを縦方向(行方向)に結合させるという方法で、データフレームfile_name_case_id_4_dfを作成する。ここで、このstr_detect()の値がTRUEにならない場合もリストfile_name_case_id_4_listの各要素にデータフレームが入っていくが、その場合は、値の何もないデータフレームが入ることになる。それらは如何せん値が無いので、bind_rows()されても縦方向に値が加わっていくことはないようだ。
# code 45
# Retrieve the cases from "file_name_case_id_2_FALSE" as possible as it is able to be.
#
file_name_case_id_4_list <- list()
#
for(i in 1:nrow(biospeciemen_6)){
#
file_name_case_id_4_list[[i]] <- file_name_case_id_2_FALSE[
str_detect(string = file_name_case_id_2_FALSE$possible_Aliquot_Submitter_ID,
pattern = biospeciemen_6$Aliquot_Submitter_ID_2[i]) == TRUE,]
#
file_name_case_id_4_list[[i]] <- file_name_case_id_4_list[[i]] %>% mutate(
ID = file_name_case_id_4_list[[i]]$ID,
Case_ID = biospeciemen_6$Case_ID[i],
Aliquot_Submitter_ID_2 = biospeciemen_6$Aliquot_Submitter_ID_2[i])}
#
names(file_name_case_id_4_list) <- biospeciemen_6$Aliquot_Submitter_ID_2
#
file_name_case_id_4_df <- bind_rows(file_name_case_id_4_list)作成したデータフレームfile_name_case_id_4_dfの列Aliquot_Submitter_ID_2で、重複するものの列Aliquot_Submitter_ID_2の値をデータフレームfile_name_case_id_4_df_overlappedに、重複しないものをデータフレームfile_name_case_id_5に入れる。このデータフレームfile_name_case_id_5の列Aliquot_Submitter_ID_2を使って、データフレームfile_name_case_id_4_dfからそれらの症例を抽出してきて、データフレームfile_name_case_id_5_uniqueを作成する。
ここで、逆にどうしてもタンパク質発現プロファイルと臨床情報を一致させることができなさそうな症例についてみるために、データフレームfile_name_case_id_4_df_overlappedの列Aliquot_Submitter_ID_2の値を使って、それらをデータフレームbiospeciemen_6から抽出し、データフレームAliquot_Submitter_ID_2_difficult_to_be_matchedを作成する。また、同様に、同定できた疾患に関してもデータフレームbiospeciemen_6から抽出し、データフレームAliquot_Submitter_ID_2_matchedを作成する。
# code 46
#
file_name_case_id_4_df_overlapped <- table(file_name_case_id_4_df$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% rename("Aliquot_Submitter_ID_2" = "Var1")
nrow(file_name_case_id_4_df_overlapped) # 482 cases
# Use "file_name_case_id_4".
file_name_case_id_5 <- table(file_name_case_id_4_df$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq == 1) %>% rename("Aliquot_Submitter_ID_2" = "Var1")
nrow(file_name_case_id_5) # 501 cases
file_name_case_id_5_unique <- file_name_case_id_4_df[file_name_case_id_4_df$Aliquot_Submitter_ID_2 %in% file_name_case_id_5$Aliquot_Submitter_ID_2,]
nrow(file_name_case_id_5_unique) # 501 cases
#
Aliquot_Submitter_ID_2_difficult_to_be_matched <- biospeciemen_6[biospeciemen_6$Aliquot_Submitter_ID_2 %in% file_name_case_id_4_df_overlapped$Aliquot_Submitter_ID_2,]
# "table_test_list_df_overlapped" summarized number of case that was not able to be extracted as unique one, because the file name did not contains the strings that is same as that in "Aliquot_Submitter_ID_2", or the strings in "Aliquot_Submitter_ID_2" is same too short.
table(file_name_case_id_5_unique$Aliquot_Submitter_ID_2 %in% file_name_case_id_5$Aliquot_Submitter_ID_2)
# TRUE
# 501
#
Aliquot_Submitter_ID_2_matched <- biospeciemen_6[biospeciemen_6$Aliquot_Submitter_ID_2 %in% file_name_case_id_5_unique$Aliquot_Submitter_ID_2,]
nrow(Aliquot_Submitter_ID_2_matched) # 501 cases
table(Aliquot_Submitter_ID_2_difficult_to_be_matched$Project_Name)データフレームfile_name_case_id_3にはデータフレームbiospeciemen_6の列Aliquot_Submitter_ID_2の値が一致するタンパク質発現プロファイル名が入っている。そのデータフレームfile_name_case_id_3の列nameの値と、code 46で作成したデータフレームfile_name_case_id_5_uniqueの列nameの値が重複していないことを確認すると、ちゃんとFALSEだけだった。
# code 47
# file_name_case_id_5
# file_name_case_id_7_unique
dim(file_name_case_id_3) # 2046 6
dim(file_name_case_id_5_unique) # 477 6
colnames(file_name_case_id_3)
table(colnames(file_name_case_id_3) %in% colnames(file_name_case_id_5_unique))
# TRUE
# 6
table(file_name_case_id_3$name %in% file_name_case_id_5_unique$name)
# FALSE
# 2046
code 48では、データフレームfile_name_case_id_3にある列name、列ID、possible_Aliquot_Submitter_ID、列PDC_Study_ID、列Case_ID、列Aliquot_Submitter_ID_2の値で、どのくらい重複しているのかを確認する。PDC_Study_IDにも重複があるが、同じプロジェクト内で複数の症例があることは当然なのでこれは問題ない。列Case_IDに481例も重複していることが問題と言える。これはどんな症例が重複しているのか確認する必要がある。このために、重複している列Case_IDの値とその頻度をデータフレームcheck_file_name_case_id_3_Case_IDとして作成した。
# code 48
biospeciemen_6
clinical_info_merged
table(file_name_case_id_3$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_3$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_3$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_3$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 14
table(file_name_case_id_3$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 481
table(file_name_case_id_3$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
check_file_name_case_id_3_name <- table(file_name_case_id_3$name) %>% data.frame() %>% filter(Freq > 1) %>% rename("name" = "Var1")
check_file_name_case_id_3_ID <- table(file_name_case_id_3$ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("ID" = "Var1")
check_file_name_case_id_3_possible_Aliquot_Submitter_ID <- table(file_name_case_id_3$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("possible_Aliquot_Submitter_ID" = "Var1")
check_file_name_case_id_3_Case_ID <- table(file_name_case_id_3$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")データフレームcheck_file_name_case_id_3_Case_IDの列Case_IDを使って、それはどんな症例なのかを確認するために、データフレームfile_name_case_id_3は、タンパク質発現プロファイル名が、biospeciemenのAliquot Submitter IDと一致している例であり、これはデータフレームclinicalやデータフレームfollowupと結合しても大丈夫なはずの症例である。その結合に用いるのが列Case_IDであり、これが重複しているのはけっこう不味い。
なので、code 48で作成したデータフレームcheck_file_name_case_id_3_Case_IDの列Case_IDを使って、データフレームfile_name_case_id_3から列Case_IDの値が重複している症例を抽出し、データフレームfile_name_case_id_3_1を作成する。それを見てみると、同じ症例から取得された別のタンパク質発現プロファイルのようだ。例えば、正常組織とか、採取するタイミングとかが違っているらしい。code 49の10行目以降のtable()でそれがわかる。
別の組織や別のタイミングで採取されたタンパク質発現プロファイルに、同じ症例の情報が登録されているのは、現時点では問題ないように思う。ただし、タイミングはちょっと不味いかもしれない。例えば、ある肺がん患者由来の腫瘍組織を時期を変えて採材してタンパク質発現プロファイルを取得した、ということである。その場合、疾患ごとのN数に重複が生じてしまうじゃあないか。これはあとから考えよう。
# code 49
file_name_case_id_3_1 <- file_name_case_id_3[file_name_case_id_3$Case_ID %in% check_file_name_case_id_3_Case_ID$Case_ID == TRUE,]
# several cases are from same patient. These are normal or tumor tissues, and probably the timing of harvested was different.
# example; Case_ID; f1ed9eb2-cf1e-11e9-9a07-0a80fada099c
clinical_info_merged[clinical_info_merged$Case_ID == "f1ed9eb2-cf1e-11e9-9a07-0a80fada099c",]$Sample_Type
clinical_info_merged[clinical_info_merged$Case_ID == "f1ed9eb2-cf1e-11e9-9a07-0a80fada099c",]$Tissue_Type
clinical_info_merged[clinical_info_merged$Case_ID == "f1ed9eb2-cf1e-11e9-9a07-0a80fada099c",]$Days_to_Last_Follow_Up次に、code 46で作成したデータフレームfile_name_case_id_5_uniqueでも、列name、列ID、possible_Aliquot_Submitter_ID、列PDC_Study_ID、列Case_ID、列Aliquot_Submitter_ID_2の値で、どのくらい重複しているのかを確認する。
やっぱり、列Case_IDに重複した値があるようだ。さらに、こちらは更に問題で、列nameや列IDにまで重複がある。列nameは、タンパク質発現プロファイルの列名、列IDは元のデータフレームに1から順番に振った番号、列possible_Aliquot_Submitter_IDはタンパク質発現プロファイル名から、Aliquot Submitter IDに使用されているであろう部分を取ってきた値である。これが重複しているということは、別の症例に、別のタンパク質発現プロファイル名が割り当てられている可能性があるということだ。これは不味い。なので、code 48と同様に、重複した値が確認された値をデータフレームcheck_file_name_case_id_5_unique_name、check_file_name_case_id_5_unique_ID、possible_Aliquot_Submitter_ID、check_file_name_case_id_5_unique_Case_IDを作成する。
# code 50
table(file_name_case_id_5_unique$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 25
table(file_name_case_id_5_unique$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 25
table(file_name_case_id_5_unique$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 25
table(file_name_case_id_5_unique$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 10
table(file_name_case_id_5_unique$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 83
table(file_name_case_id_5_unique$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
check_file_name_case_id_5_unique_name <- table(file_name_case_id_5_unique$name) %>% data.frame() %>% filter(Freq > 1) %>% rename("name" = "Var1")
check_file_name_case_id_5_unique_ID <- table(file_name_case_id_5_unique$ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("ID" = "Var1")
check_file_name_case_id_5_unique_possible_Aliquot_Submitter_ID <- table(file_name_case_id_5_unique$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("possible_Aliquot_Submitter_ID" = "Var1")
check_file_name_case_id_5_unique_Case_ID <- table(file_name_case_id_5_unique$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")code 50で作成したデータフレームのうち、自分が最も不味いと思うデータフレームcheck_file_name_case_id_5_unique_Case_IDとデータフレームcheck_file_name_case_id_5_unique_nameを使って、それらが一体どんな症例だったのかを抽出してくる。それがデータフレームfile_name_case_id_5_unique_Case_ID_duplicatedとデータフレームfile_name_case_id_5_unique_name_duplicatedである。
行7から9,行18から21で、それらはどんな症例なのかを見ているが、そこでわかったことは、やはり、これらは一つのタンパク質発現プロファイルに複数の臨床情報が関連付けられている可能性があることだ。これらはbiospeciemenを見ている限りでは、列Aliquot IDで同定は出来るのだが、その列Aliquote IDがタンパク質発現プロファイルに関連していない。そして、自分にはそれを分離するための情報を見つけることが出来なかった。
依って、データフレームfile_name_case_id_5_unique_name_duplicatedの列nameが重複しているような症例は解析から除き、データフレームfile_name_case_id_6_uniqueを作成する。
# code 51
file_name_case_id_5_unique_Case_ID_duplicated <- file_name_case_id_5_unique[file_name_case_id_5_unique$Case_ID %in% check_file_name_case_id_5_unique_Case_ID$Case_ID == TRUE,]
# example; Case_ID; 338845cd-4ec7-404e-89b5-eecd05c9f3e5
clinical_info_merged[clinical_info_merged$Case_ID == "338845cd-4ec7-404e-89b5-eecd05c9f3e5",]$Sample_Type
clinical_info_merged[clinical_info_merged$Case_ID == "338845cd-4ec7-404e-89b5-eecd05c9f3e5",]$Tissue_Type
clinical_info_merged[clinical_info_merged$Case_ID == "338845cd-4ec7-404e-89b5-eecd05c9f3e5",]$Days_to_Last_Follow_Up
# Probably, name duplication is problematic.
file_name_case_id_5_unique_name_duplicated <- file_name_case_id_5_unique[file_name_case_id_5_unique$name %in% check_file_name_case_id_5_unique_name$name == TRUE,]
check_file_name_case_id_5_unique_name_duplicated_Case_ID <- table(file_name_case_id_5_unique_name_duplicated$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")
# example; Case_ID; e8d578a1-693a-11ea-b1fd-0aad30af8a83
clinical_info_merged[clinical_info_merged$Case_ID == "e8d578a1-693a-11ea-b1fd-0aad30af8a83",]$Sample_Type
clinical_info_merged[clinical_info_merged$Case_ID == "e8d578a1-693a-11ea-b1fd-0aad30af8a83",]$Tissue_Type
clinical_info_merged[clinical_info_merged$Case_ID == "e8d578a1-693a-11ea-b1fd-0aad30af8a83",]$Days_to_Last_Follow_Up
check_clinical_info_merged_temp <- clinical_info_merged[clinical_info_merged$Case_ID == "e8d578a1-693a-11ea-b1fd-0aad30af8a83",]
# IMPORTANT NOTICE!!!!
# For example, the Case_ID == "e8d578a1-693a-11ea-b1fd-0aad30af8a83" looks problem, "Aliquot_Submitter_ID", "Tissue_Type" are different but "name" (file name of protein intensity), "Case_ID" are same. It means each samples (or aliquote of case) are from Tumor or Normal tissue, but mass spec data is shared or same. If the aliquot is from different tissue, the mass spec data should be saparated, but the case is not. Since the Case_ID == "e8d578a1-693a-11ea-b1fd-0aad30af8a83" ("name" == "PDC000214_Log_T90_N89"), and all similar cases are hard to distinguish between Tumor or Normal tissue, these case should be removed from further analysis. THESE SAMPLES ARE POOLED SAMPLE WITH TUMOR AND NORMAL TISSUE.
# IMPORTANT NOTICE!!!!
# Proteome results from "Human Early-Onset Gastric Cancer - Korea University" is shit. Remove them (file_name_case_id_5_unique_name_duplicated$name) from further analysis.
check_clinical_info_merged_temp <- clinical_info_merged[clinical_info_merged$Case_ID %in% file_name_case_id_5_unique_name_duplicated$Case_ID == TRUE,]
# Use following data frame.
file_name_case_id_6_unique <- file_name_case_id_5_unique[file_name_case_id_5_unique$name %in% file_name_case_id_5_unique_name_duplicated$name == FALSE,]code 51の最後に作成した、一つのタンパク質発現プロファイルに複数のIDが割り振られている例(実際は同じ患者の正常組織と腫瘍組織をプールした例であるが、こんなものは正常組織なのか腫瘍組織なのかわからない)を集めたデータフレームfile_name_case_id_6_uniqueの列name、列ID、列possible_Aliquot_Submitter_ID、列PDC_Study_ID、列Case_ID、列Aliquot_Submitter_ID_2の重複を確認する。列Case_IDの重複は同じ患者由来の別組織のタンパク質発現プロファイルを意味しており、それらがある可能性はあるので、それはここでは許容する。
# code 52
table(file_name_case_id_6_unique$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_6_unique$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_6_unique$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_6_unique$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_6_unique$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 58
table(file_name_case_id_6_unique$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
check_file_name_case_id_6_unique_name <- table(file_name_case_id_6_unique$name) %>% data.frame() %>% filter(Freq > 1) %>% rename("name" = "Var1")
check_file_name_case_id_6_unique_ID <- table(file_name_case_id_6_unique$ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("ID" = "Var1")
check_file_name_case_id_6_unique_unique_possible_Aliquot_Submitter_ID <- table(file_name_case_id_6_unique$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("possible_Aliquot_Submitter_ID" = "Var1")
check_file_name_case_id_5_unique_Case_ID <- table(file_name_case_id_6_unique$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")
# Example; Case_ID; e544fb5a-5b53-4e1d-a3ab-e80239baca87
clinical_info_merged[clinical_info_merged$Case_ID == "e544fb5a-5b53-4e1d-a3ab-e80239baca87",]$Sample_Type
clinical_info_merged[clinical_info_merged$Case_ID == "e544fb5a-5b53-4e1d-a3ab-e80239baca87",]$Tissue_Type
clinical_info_merged[clinical_info_merged$Case_ID == "e544fb5a-5b53-4e1d-a3ab-e80239baca87",]$Days_to_Last_Follow_Up
file_name_case_id_6_unique[file_name_case_id_6_unique$Case_ID == "e544fb5a-5b53-4e1d-a3ab-e80239baca87",] %>% view()code 53では、データフレームfile_name_case_id_3とデータフレームfile_name_case_id_6_uniqueの列name(タンパク質発現プロファイル名)やその他の列にに重複がないことを確認して、最後にそれらをbind_rows()で縦方向(行方向)に結合して、データフレームfile_name_case_id_7を作成する。最終的に2523例を生存期間解析に使用することができそう。
# code 53
# All file_name are identical, so merge them.
# Check duplicated observations.
table(file_name_case_id_3$name %in% file_name_case_id_6_unique$name)
# FALSE
# 2046
# check_file_name_case_id_3 <- file_name_case_id_3[file_name_case_id_3$Case_ID %in% file_name_case_id_5_unique$Case_ID == TRUE,]
# check_file_name_case_id_5_unique <- file_name_case_id_5_unique[file_name_case_id_5_unique$Case_ID %in% file_name_case_id_3$Case_ID == TRUE,]
table(file_name_case_id_3$ID %in% file_name_case_id_6_unique$ID)
# FALSE
# 2046
table(file_name_case_id_3$possible_Aliquot_Submitter_ID %in% file_name_case_id_6_unique$possible_Aliquot_Submitter_ID)
# FALSE
# 2046
table(file_name_case_id_3$Case_ID %in% file_name_case_id_6_unique$Case_ID)
# FALSE
# 2046
table(file_name_case_id_3$Aliquot_Submitter_ID_2 %in% file_name_case_id_6_unique$Aliquot_Submitter_ID_2)
# FALSE
# 2046
file_name_case_id_7 <- bind_rows(file_name_case_id_3, file_name_case_id_6_unique)
dim(file_name_case_id_6_unique) # 2523 6code 54で、タンパク質発現プロファイル名と、bispeciemenの情報を結びつけることが出来そうな症例で構成されたデータフレームfile_name_case_id_7を作成したが、それらの重複した値を上記と同様の方法で確認していく。列PDC_Study_IDと列Case_IDの重複はあり得るのでここでは許容する。
# code 54
table(file_name_case_id_7$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 23 study
table(file_name_case_id_7$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 539 sample is from same patients, but file name will be different.
table(file_name_case_id_7$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
check_name <- table(file_name_case_id_7$name) %>% data.frame() %>% filter(Freq > 1) %>% rename("name" = "Var1")
check_ID <- table(file_name_case_id_7$ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("ID" = "Var1")
check_possible_Aliquot_Submitter_ID <- table(file_name_case_id_7$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("possible_Aliquot_Submitter_ID" = "Var1")
check_Case_ID <- table(file_name_case_id_7$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")確認が終わって大丈夫そうなので、データフレームfile_name_case_id_7をwrite_tsv()でTSVファイルとして出力しておく。
# code 55
write_tsv(file_name_case_id_7, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/file_name_case_id_matching.tsv") # for file name and Case_ID matching.データフレームfile_name_case_id_7は、実質的には「解析に使えそうなタンパク質発現プロファイル名」しか情報が無いので、それを使って、一致する症例を抽出してくる。code 56ではまずはbiospeciemenから抽出する。必要な処理は18行目からであり、5から14行目まではtable()による確認のみである。データフレームbiospeciemen_6には、解析に使えそうな症例やサンプルの情報が記載されている。そこから、データフレームfile_name_case_id_7の列Case_IDが一致している症例を抽出してきて、データフレームbiospeciemen_7を作成する。この後、データフレームを作成するごとにnrow()で行数、すなわち症例数を確認しながら進める。続いて、データフレームbiospeciemen_7からデータフレームfile_name_case_id_7の列Aliquot_Submitter_ID_2と一致する症例をさらに抽出してデータフレームbiospeciemen_8とし、nrow()で症例数を確認する。そして、このデータフレームbiospeciemen_8の列Aliquot_Submitter_ID_2(この値の重複は困る)や列Case_ID(この値の重複は有り得る)に重複があるかどうかも確認する。そしてデータフレームbiospeciemen_8と、タンパク質発現プロファイルの列名を関連付けることになるデータフレームfile_name_case_id_7をinner_join()で結合し、データフレームclinical_info_analysisを作成する。この時点で2473例が残った。
# code 56
# Merge file_name_case_id_7 with clinical information
nrow(file_name_case_id_7) # 2473 cases
colnames(file_name_case_id_7)
table(file_name_case_id_7$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 23 study
table(file_name_case_id_7$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 539 sample is from same patients, but file name will be different.
table(file_name_case_id_7$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(biospeciemen_6$Aliquot_Submitter_ID %in% biospeciemen_6$Aliquot_Submitter_ID_2)
# biospeciemen
biospeciemen_7 <- biospeciemen_6[biospeciemen_6$Case_ID %in% file_name_case_id_7$Case_ID == TRUE,]
nrow(biospeciemen_7) # 2495 cases
biospeciemen_8 <- biospeciemen_7[biospeciemen_7$Aliquot_Submitter_ID_2 %in% file_name_case_id_7$Aliquot_Submitter_ID_2 == TRUE,]
nrow(biospeciemen_8) # 2473 cases
table(biospeciemen_8$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(biospeciemen_8$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 539 sample is from same patients
clinical_info_analysis <- inner_join(file_name_case_id_7, biospeciemen_8, by = c("Case_ID", "Aliquot_Submitter_ID_2"), suffix = c("", "_biospecimen"))
nrow(clinical_info_analysis) # 2473 cases
# check_file <- file_name_case_id_7[file_name_case_id_7$Case_ID %in% clinical_2$Case_ID == TRUE,]
table(clinical_info_analysis$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 539
上記のcode 56で作成したデータフレームclinical_info_analysisの列Case_IDと一致する症例をデータフレームclinicalから抽出してきて、データフレームclinical_2を作成する。ここでも、データフレームclinical_2の列Case_IDの値が重複している症例は何なのか確認する。すると、どうしても列Case_IDで重複してしまっている例がある。code 55まではタンパク質発現プロファイルの列名に関する処理だったが、これはデータフレームclinicalに関する処理で別であり、依って、ここでも重複は逐一確認すべきと思う。データフレームclinical_2の列Case_IDで重複している値をデータフレームclinical_2_duplicated_Case_IDとし、それを使ってデータフレームclinical_2から、重複している列Case_IDの値を持つ症例を抽出し、データフレームclinical_2_duplicatedを作成する。
データフレームclinical_2_duplicatedを見ていると、列Annotationに「the pooled sample」という記載のある症例がある。なので、これらをstr_detect()で除き、データフレームclinical_2_remove_pooledを作成する。このデータフレームclinical_2_remove_pooledは、このオブジェクト名通り、複数の検体をプールせずに解析された症例の臨床情報である。これをデータフレームclinical_2と統合するひつようがあるので、まずはデータフレームclinical_2から、データフレームclinical_2_duplicatedの列Case_IDを持つ症例を全部除き、それをデータフレームclinical_2_1を作成し、そのデータフレームclinical_2_1とデータフレームclinical_2_remove_pooledを縦方向(行方向)に結合することでデータフレームclinical_3、すなわち、解析できそうな症例のうち、プールされた検体が使用されている症例を除いたものを作成する。
このデータフレームclinical_3の列Case_IDの重複を調べると、未だに2例ずつ重複していることがわかる。これらはどんなタンパク質発現プロファイルだろうと思って、データフレームfile_name_case_id_7からそれらを抽出してデータフレームcheck_fileを作成し、それを見てみた(code 57の37から45行目)。しかし、どうやらタンパク質発現プロファイルの列名には重複がなかった。つまり、1つのタンパク質発現プロファイルに、複数のCase_IDが割り振られているらしい。これは言うてみれば、複数の症例(患者)から、同じタンパク質発現プロファイルが取得されたということである….あかんやん。そんなわけないやん。なんで別患者なのにタンパク質発現プロファイル一緒やねん。なので、データフレームclinical_3から、データフレームclinical_3_duplicated_Case_IDの列Case_IDを持つ症例を除外し、データフレームclinical_4を作成する。このデータフレームclinical_4の列Case_IDは、重複は内容だった(58行目)。
なので、code 56で作成したデータフレームclinical_info_analysisと、ここで作成したデータフレームclinical_4をfull_join()する。full_join()するとby = で指定された列に同じ値がない場合は、その行はNAで結合できる。inner_join()とかだと、by=で指定された列に同じものがない場合はそれはデータフレームから削除されてしまう。以降の65から75行目まではtable()による列の値の確認である。
# code 57
# clinical
colnames(clinical)
clinical_2 <- clinical[clinical$Case_ID %in% clinical_info_analysis$Case_ID == TRUE,]
dim(clinical_2) # [1] 1949 152
table(clinical_2$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 38
clinical_2_duplicated_Case_ID <- table(clinical_2$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")
clinical_2[clinical_2$Case_ID == "6c3a122e-9f69-4252-bbaa-3ee501db83c1",] %>% view()
clinical_2_duplicated <- clinical_2[clinical_2$Case_ID %in% clinical_2_duplicated_Case_ID$Case_ID == TRUE,]
# several cases are pooled sample. Why do they connect the clinical information of pooled sample into that of unique patient!? REMOVE THEM.
# clinical_2_remove_pooled <- clinical_2_duplicated[str_detect(string = clinical_2_duplicated$Annotation, pattern = "pool") == FALSE,]
# test <- clinical_2[str_detect(string = clinical_2_duplicated$Annotation, pattern = "pool") == TRUE,] # it can not be worked
clinical_2_remove_pooled <- clinical_2 %>% filter(str_detect(string = clinical_2$Annotation, pattern = "pool") == FALSE)
# table(clinical_2_remove_pooled$Case_ID %in% test$Case_ID)
# TRUE
# 64
# removed pooled sample;
# clinical_2_1; remove cases with Case_ID duplication
# clinical_2_remove_pooled; remove pooled sample from clinical_2_1
# clinical_3; removed pooled sample from clinical_2_1 and add them into clinical_2_1
clinical_2_1 <- clinical_2[clinical_2$Case_ID %in% clinical_2_duplicated$Case_ID == FALSE,] # extract identical Case_ID
clinical_3 <- bind_rows(clinical_2_1, clinical_2_remove_pooled)
table(clinical_3$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 28
check_clinical_3_Case_ID <- table(clinical_3$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")
clinical_3_duplicated_Case_ID <- clinical_3[clinical_3$Case_ID %in% check_clinical_3_Case_ID$Case_ID == TRUE,]
# What are these cases??
# file_name_case_id_7$name vs clinical_3_duplicated_Case_ID$Case_ID
check_file <- file_name_case_id_7[file_name_case_id_7$Case_ID %in% clinical_3_duplicated_Case_ID$Case_ID == TRUE,]
table(check_file$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
# file_name_case_id_7$name vs clinical_3$Case_ID
check_file <- file_name_case_id_7[file_name_case_id_7$Case_ID %in% clinical_3$Case_ID == TRUE,]
table(check_file$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
# file_name_case_id_7$name vs clinical_2$Case_ID
check_file <- file_name_case_id_7[file_name_case_id_7$Case_ID %in% clinical_2$Case_ID == TRUE,]
table(check_file$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
# clinical_3 file names are identical, but clinical information was doubled. REMOVE THEM because accurate matching between file name and Case_ID is difficult.
clinical_4 <-
clinical_3[clinical_3$Case_ID %in% clinical_3_duplicated_Case_ID$Case_ID == FALSE,] # remove duplicated Case_ID
table(clinical_4$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
dim(clinical_4) # [1] 1879 152
colnames(clinical_4)
check_file <- file_name_case_id_7[file_name_case_id_7$Case_ID %in% clinical_4$Case_ID == TRUE,]
table(check_file$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
clinical_info_analysis <- full_join(clinical_info_analysis, clinical_4, by = c("Case_ID", "Case_Submitter_ID"), suffix = c("", "_clinical"))
dim(clinical_info_analysis) # [1] 2473 200
table(clinical_info_analysis$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 23 study
table(clinical_info_analysis$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 589 sample is from same patients, but file name will be different.
table(clinical_info_analysis$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0次に、データフレームfollowupを整理する。その前に、データフレームtreatmentとデータフレームexposureについて述べる。データフレームtreatmentやデータフレームexposureには、例えば投与された抗がん剤の種類や放射線治療の有無などの情報が入っており、これを1つの症例に関連しようとすると、オブザベーションの数(ここで言うところの列数)が膨れ上がる。その理由は、当然のように一人の患者に複数のモダリティーが使用されるためである。なので、上述の通り、この解析にはtreatmentとexposureは入れないことにする。もしこれらの情報を加味した生存期間解析を行う場合は、少し趣向の違う解析になりそうである。
データフレームtreatmentやデータフレームexposureと違って、データフレームfollowupには、例えば、列Days_To_Follow_Upなんかが含まれており、全生存期間の評価に使う可能性もある。なので、必要である。そこで、データフレームfollowupから、code 57で作成したデータフレームclinical_info_analysisの列Case_IDを持つ症例を抽出してデータフレームfollowup_2を作成する。ところが、解析出来そうな症例でこのデータフレームfollowupの情報を持つ症例は227例しかないらしい。しゃあないので、これらは後ほどfull_join()しようと思う。
データフレームfollowup_2は少し忘れて、生存期間解析に必要な列を取って来ようと思う。42行目からがそのコードである。その必要と思われる(実際には使わないものもある)列をベクトルcolumns_for_survivalに保存する。これらがこの解析で含めた列であるが、これはデータフレームbioscpeciemenかデータフレームfollowupのどちらにあるのかを把握しておく。まず、ベクトルcolumns_for_survivalと一致する列、しない列をデータフレームclinical_info_analysis(これはデータフレームbiospeciemenとデータフレームclinicalが結合されている)の列名から取ってきて、一致する値をベクトルcolumns_clinical_info_analysis、一致しない値をベクトルcolumns_other_than_biospeciemen_clinicalとする。
# code 58
# treatment
table(file_name_case_id_7$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(file_name_case_id_7$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
treatment_2 <- treatment[treatment$Case_ID %in% clinical_info_analysis$Case_ID == TRUE,]
dim(treatment_2) # [1] 84 2
table(treatment_2$Case_ID %in% clinical_info_analysis$Case_ID)
# TRUE
# 84
colnames(treatment_2)
# exposure
exposure_2 <- exposure[exposure$Case_ID %in% clinical_info_analysis$Case_ID == TRUE,]
dim(exposure_2) # [1] 1162 33
table(exposure_2$Case_ID %in% clinical_info_analysis$Case_ID)
# TRUE
# 1162
colnames(exposure_2)
# followup
followup_2 <- followup[followup$Case_ID %in% clinical_info_analysis$Case_ID == TRUE,]
dim(followup_2) # [1] 227 70
table(followup_2$Case_ID %in% clinical_info_analysis$Case_ID)
# TRUE
# 227
table(followup_2$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0, There is no overlapped information.
colnames(followup_2)
columns_for_survival <- c("Case_ID",
"Case_Submitter_ID",
"Days_To_Follow_Up",
"Status",
"Cause_of_Death",
"Vital_Status",
"Days_To_Progression",
"Days_To_Recurrence",
"Days_To_Progression_Free",
"Disease_Response",
"Days_to_Best_Overall_Response",
"Days_to_Diagnosis",
"Days_to_New_Event",
"Days_to_Diagnosis",
"Progression_Free_Survival",
"Progression_Free_Survival_Event",
"Residual_Disease",
"Days_to_Death",
"Days_to_Last_Follow_Up",
"Progression_or_Recurrence",
"Last_Known_Disease_Status",
"Primary_Diagnosis",
"Site_of_Resection_or_Biopsy",
"Tissue_or_Organ_of_Origin",
"Primary_Site",
"Disease_Type")
length(columns_for_survival) # 26 columns
columns_clinical_info_analysis <- columns_for_survival[columns_for_survival %in% colnames(clinical_info_analysis) == TRUE]
length(columns_clinical_info_analysis) # 21 columns
columns_clinical_info_analysis
# [1] "Case_ID" "Case_Submitter_ID" "Status" "Cause_of_Death"
# [5] "Vital_Status" "Days_to_Best_Overall_Response" "Days_to_Diagnosis" "Days_to_New_Event"
# [9] "Days_to_Diagnosis" "Progression_Free_Survival" "Progression_Free_Survival_Event" "Residual_Disease"
# [13] "Days_to_Death" "Days_to_Last_Follow_Up" "Progression_or_Recurrence" "Last_Known_Disease_Status"
# [17] "Primary_Diagnosis" "Site_of_Resection_or_Biopsy" "Tissue_or_Organ_of_Origin" "Primary_Site"
# [21] "Disease_Type"
columns_other_than_biospeciemen_clinical <- columns_for_survival[columns_for_survival %in% colnames(clinical_info_analysis) == FALSE]
length(columns_other_than_biospeciemen_clinical) # 5 columns
columns_other_than_biospeciemen_clinical
# [1] "Days_To_Follow_Up" "Days_To_Progression" "Days_To_Recurrence" "Days_To_Progression_Free" "Disease_Response"
followup_2[,colnames(followup_2) %in% columns_other_than_biospeciemen_clinical == TRUE] %>% view()code 59で、データフレームclinical_info_analysisとデータフレームfollowup_2をfull_join()し、それを改めてデータフレームclinical_info_analysisとする。12から23行目まで、これまで行ってきたように列Case_IDや列nameなどの重複を確認し、問題なさそうなことを確認する。そして、列nameの値は症例を識別するには長過ぎるので、データフレームclinical_info_analysisに新しく列ID_2をmutate()で作り、そこに文字列「case」と列IDの値を繋げた、例えばcase1323のような値を入れていく。出来上がったデータフレームclinical_info_analysisは、後ほど生存期間解析に使用するので、write_tsv()でTSVファイルとして外部に保存する。
# code 59
# clinical_info_analysis <- full_join(clinical_info_analysis, treatment_2, by = c("Case_ID", "Case_Submitter_ID"), suffix = c("", "_treatment"))
# dim(clinical_info_analysis) # [1] 2575 225
# clinical_info_analysis <- full_join(clinical_info_analysis, exposure_2, by = c("Case_ID", "Case_Submitter_ID"), suffix = c("", "exposure"))
# dim(clinical_info_analysis) # [1] 2575 257
clinical_info_analysis <- full_join(clinical_info_analysis, followup_2, by = c("Case_ID", "Case_Submitter_ID"), suffix = c("", "followup"))
dim(clinical_info_analysis) # [1] 2473 268
table(clinical_info_analysis$name) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$possible_Aliquot_Submitter_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(clinical_info_analysis$PDC_Study_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 23 study
table(clinical_info_analysis$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 589 sample is from same patients, but file name will be different.
table(clinical_info_analysis$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
check_name <- table(clinical_info_analysis$name) %>% data.frame() %>% filter(Freq > 1) %>% rename("name" = "Var1")
check_ID <- table(clinical_info_analysis$ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("ID" = "Var1")
check_Aliquot_Submitter_ID_2 <- table(clinical_info_analysis$Aliquot_Submitter_ID_2) %>% data.frame() %>% filter(Freq > 1) %>% rename("Aliquot_Submitter_ID_2" = "Var1")
check_Case_ID <- table(clinical_info_analysis$Case_ID) %>% data.frame() %>% filter(Freq > 1) %>% rename("Case_ID" = "Var1")
check_duplicated <- clinical_info_analysis[clinical_info_analysis$name %in% check_name$name == TRUE,] # nothing. It looks OK!
#
clinical_info_analysis <- clinical_info_analysis %>% mutate(
ID_2 = paste0("case", ID)
)
write_tsv(clinical_info_analysis, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/clinical_info_analysis.tsv") # for file name and Case_ID matching.
dim(clinical_info_analysis) # [1] 2473 269生存期間解析に使用出来る症例から構成されるデータフレームclinical_info_analysisの列nameと一致するタンパク質発現プロファイル名を、データフレームresult_missForest_pre_argu_df_median_normalized_df(missForestで欠損値を補完し、かつ、CPTACが計算した中央値に近い値を得ることができたデータフレーム)から選んできて、データフレームresult_missForest_pre_argu_df_median_normalized_df_2を作成する。それらを転置したり行名を付けたりして、データフレームclinical_info_analysisとinner_join()出来る形に整え、最終的にデータフレームanalysisを作成する。このデータフレームanalysisは生存期間解析で使用するので、write_tsv()で外部にTSVファイルとして保存しておく。このデータは、後ほど生存期間解析のために使用する。
# code 60
result_missForest_pre_argu_df_median_normalized_df_2 <-
result_missForest_pre_argu_df_median_normalized_df[, colnames(result_missForest_pre_argu_df_median_normalized_df) %in% clinical_info_analysis$name == TRUE]
dim(result_missForest_pre_argu_df_median_normalized_df_2)
# [1] 5208 2473
result_missForest_pre_argu_df_median_normalized_df_2_t <- t(result_missForest_pre_argu_df_median_normalized_df_2) %>% data.frame()
result_missForest_pre_argu_df_median_normalized_df_3 <- result_missForest_pre_argu_df_median_normalized_df_2_t %>% rownames_to_column("name")
dim(result_missForest_pre_argu_df_median_normalized_df_3)
# 2473 5209; 5208 genes + column "name" = 5209
analysis <- inner_join(clinical_info_analysis, result_missForest_pre_argu_df_median_normalized_df_3, by = "name")
table(analysis$ID) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
table(analysis$ID_2) %>% data.frame() %>% filter(Freq > 1) %>% nrow() # 0
write_tsv(analysis, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/analysis.tsv") # This is for further analysis.上記で臨床情報とタンパク質発現プロファイルは結合することが出来た。ここまでの解析をRDataとして保存しておく。
# code 61
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_4.RData")escape、gsva、fgseaを使ってGSEA(Gene Set Enrichment Analysis)を行う
このRのコードでは、次にタンパク質と遺伝子発現の相関をとるためデータの整理に入るが、これも随分大変でコード自体も結構長い。CPTACのタンパク質発現プロファイルとTCGAの遺伝子発現プロファイルのマッチングをしているうちに、ここまで行ったデータが一体どんなものだったのか、絶対にわすれてしまう。だからと言って、この相関の解析を他の記事にまとめるのも非常に面倒くさい。この記事のCode 42まで同じようなデータ整理を行う羽目になる。なので、先にGSEA(Gene Set Enrichment Analysis)と生存期間期間解析を行うことにする。code 97に飛ぶ。ファイルは以下である。
ここから使用するRのダウンロード2
読むのが面倒な人は、コードを読んだら良いと思う。コードはここで販売している。ZIPを解凍するとRのコードがいくつかあるので、そのうちの「2 CPTAC GSEA enrichment survival.Rmd」というファイルが、以降のコードである。
code 61までの解析をRStudioで行ってる場合、それを閉じてしまって良い。そうしなければメモリに使わないオブジェクトをメモリに保存したままになって、データのやりとりがかなり遅くなってしまう。code 97では、新しくRStudioを開き直して、改めて同じようなパッケージをlibrary()で読んで、code 61で外部に保存したanalysis.tsvをfread()で読み込んでくる。
# code 97
## Read the data frame, that was combined with clinical information and protein expression
analysis <- fread(
input = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/analysis.tsv",
sep = "\t",
encoding = "UTF-8",
data.table = FALSE) # This data must be used for the analysis
データフレームanalysisは、タンパク質発現プロファイルの列だけでなく、他の列も色々含んでいる。各種GSEAに必要な列は269列目の列ID_2からなので、それ以降を選んできてデータフレームanalysis_2を作成し、列ID_2をデータフレームanalysis_2の行名にする。この状態では行が各症例もしくは各サンプルのタンパク質発現プロファイル(ここではこれをタンパク質発現プロファイル名と呼ぶ)、列がタンパク質であり、各種GSEAが処理することができない。なので、t()で転置させて、マトリックスanalysis_2_tを作成する。また、この時点でデータは中央値により正規化した底2の対数なので、これを真数に直し、それをマトリックスanalysis_2_t_arguとする。そしてこのマトリックスをas.data.frame()によりデータフレームanalysis_2_t_argu_dfに直し、行名としていた値を改めて列に直し、その列名を”protein”とする。
# code 98
## Prepare matrix for GSEA.
## column 5529 is column "case" that was prepared at code 56.
## column 5 to 5212 are protein expression.
analysis_2 <- analysis[, c(269:ncol(analysis))]
analysis_2 <- analysis_2 %>% column_to_rownames(var = "ID_2")
analysis_2_t <- t(analysis_2)
analysis_2_t_argu <- 2^(analysis_2_t)
# Check dataset looks like.
view(analysis_2_t_argu[1:100, 1:100])
hist(analysis_2_t_argu[1:100, 1:100], breaks = 1000)
dim(analysis_2_t_argu) #[1] 5208 1889
# the matrix get back to data frame for preparation of GCT file
analysis_2_t_argu_df <- as.data.frame(analysis_2_t_argu)
analysis_2_t_argu_df <- analysis_2_t_argu_df %>% rownames_to_column(var = "protein")ssGSEA2
code 99では、ssGSEA2に使用するGCTファイルを作成する。しかし、今回はssGSEA2は行わない。MigSigDBに登録されている遺伝子セット全部を使ってssGSEA2を行うと、時間が掛かり過ぎるためである。ただし、ここで作成するGCTファイルとダウンロードしたGMTファイルを使ってssGSEA2はちゃんと実行出来たので、必要ならばこのコードを使用すれば良いと思う。
# code 99
## Prepare GCT file
header <- "#1.2"
number_protein <- nrow(analysis_2_t_argu_df) # 5208 proteins
number_case <- ncol(analysis_2_t_argu_df)-1 # 2473 cases
file.remove("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/CPTAC_4_4.gct")
cat(header,"\n", file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/CPTAC_4_4.gct")
cat(number_protein,"\t",number_case,"\n", file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/CPTAC_4_4.gct", append = TRUE)
write_tsv(analysis_2_t_argu_df, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/CPTAC_4_4.gct",
col_names = TRUE,
append = TRUE)code 100はコード全体がコメントアウトされているが、code 99で作成したGCTファイルを使ってssGSEA2を行うコードである。これでちゃんと動くはずだが、時間が掛かり過ぎるので、今回の解析では使用しなかった。ssGSEA2は、個人のコンピューターで行う場合はおそらく300遺伝子セットくらいが限界では無いかと思う。これを行うときは、参照する遺伝子セットをある程度絞って計算を始める必要がある。サーバーとかを使っても、そんなに時間を掛けることは出来ないのでは無いかと思う。
# code 100
## 2000 gene sets were analyzed at ~20 h after starting ssGSEA2. the 36000 genesets will be spend for 18 days.
## Following code was working but not run because of too long spending time.
# start_results_ssGSEA2_NES <- Sys.time()
# set.seed(20260314)
# results_ssGSEA2_NES <- run_ssGSEA2(input.ds = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/CPTAC_4_4.gct",
# output.prefix = "NES_",
# gene.set.databases = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/msigdb.v2026.1.Hs.symbols.gmt",
# sample.norm.type = "none",
# weight = 0.75,
# correl.type = "rank",
# statistic = "area.under.RES",
# output.score.type = "NES",
# nperm = 1000,
# min.overlap = 10,
# extended.output = TRUE,
# par = 16,
# spare.cores = 0,
# global.fdr = FALSE,
# param.file=TRUE,
# log.file='run.log_NES')
# end_results_ssGSEA2_NES <- Sys.time()
gc()
gc()
# par = getOption("mc.cores",detectCores()) could be used.escape
code 101ではescapeとgsvaで使用するGMTファイルを準備する。gsvaパッケージのreadGMT()を使用する。
# code 101
# MSigDB v2026.1.Hs (Jan 2026)
# https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp
msigdb_2026_1 <- readGMT("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/msigdb.v2026.1.Hs.symbols.gmt")code 102がescapeによるエンリッチメント解析である。計算に要する時間を測定するため、開始前後の時間をSys.time()で取得し、最後に要した時間を算出する(引き算するただけ)。
# code 102
## escape, msigdb
set.seed(20260314)
start_results_escape <- Sys.time()
result_escape <- escape.matrix(
input.data = analysis_2_t_argu,
gene.sets = msigdb_2026_1,
method = "ssGSEA",
groups = 1000,
min.size = 10,
BPPARAM = MulticoreParam(workers = 16, progressbar = TRUE))
end_results_escape <- Sys.time()
gc(T,T,T)
gc(T,T,T)gvsa
code 103はgsvaによるエンリッチメント解析である。計算に要する時間を測定するため、開始前後の時間をSys.time()で取得し、最後に要した時間を算出する(引き算するただけ)。
# code 103
## gsva, msigdb
set.seed(20260314)
start_results_GSVA <- Sys.time()
gsvaPar <- gsvaParam(analysis_2_t_argu, msigdb_2026_1)
result_gsva <- gsva(gsvaPar, verbose=TRUE, BPPARAM = MulticoreParam(workers = 16, progressbar = TRUE))
end_results_GSVA <- Sys.time()
gc(T,T,T)
gc(T,T,T)fgsea
code 104はfgseaによるエンリッチメント解析である。ここではfgsea用にGMTファイルgeneset_fgseaをfgseaパッケージのgmtPathways()を使って作成する。また計算にあたってはマルチコアを使いたいので、makeCluster()で計算に使用するCPUコアを割り当てる。そして、fgseaを実行する。計算に要する時間を測定するため、開始前後の時間をSys.time()で取得し、最後に要した時間を算出する(引き算するただけ)。
# code 104
# prepare geneset for fgsea, hallmark
geneset_fgsea <- fgsea::gmtPathways("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/msigdb.v2026.1.Hs.symbols.gmt")
# From chatGPT
# choose workers (physical cores minus one is a good start)
n_workers <- detectCores(logical = FALSE)
# n_workers <- detectCores(logical = FALSE)-1
cl <- makeCluster(n_workers, type = "PSOCK") #FORK #PSOCK
registerDoParallel(cl)
# # prevent thread oversubscription inside workers (optional but recommended)
# clusterEvalQ(cl, {
# Sys.setenv(OMP_NUM_THREADS="1",
# MKL_NUM_THREADS="1",
# OPENBLAS_NUM_THREADS="1",
# RCPP_PARALLEL_NUM_THREADS="1")})
# # prevent thread oversubscription inside workers (optional but recommended)
# # this can also complete calculation.
# # fgsea was able to be run by means of adding environmental variables(su; cd /etc; gedit environment) on the /etc/environment file.
# clusterEvalQ(cl, {
# Sys.setenv(OMP_NUM_THREADS="1",
# OMP_THREAD_LIMIT="1",
# OMP_MAX_ACTIVE_LEVELS="1",
# OMP_DYNAMIC=FALSE,
# MKL_NUM_THREADS="1",
# MKL_DYNAMIC=FALSE,
# OPENBLAS_NUM_THREADS="1",
# GOTO_NUM_THREADS="1" ,
# RCPP_PARALLEL_NUM_THREADS="1")})
start_results_fgsea <- Sys.time()
set.seed(20260314)
result_fgsea <- list()
result_fgsea <- foreach(i = seq_len(ncol(analysis_2_t_argu)), .packages = "fgsea", .inorder = TRUE, .verbose = TRUE) %dopar% {
fgsea(pathways = geneset_fgsea,
stats = analysis_2_t_argu[, i],
minSize = 10,
maxSize = 500)} # %dopar% %do%
# n <- 100
# result_fgsea <- foreach(i = c(1:n), .packages = "fgsea", .inorder = TRUE, .verbose = TRUE) %dopar% {
# fgsea(pathways = geneset_fgsea,
# stats = analysis_2_t_argu[, i],
# minSize = 10,
# maxSize = 500)} # %dopar% %do%
stopCluster(cl)
rm(cl)
# foreach::registerDoSEQ()
end_results_fgsea <- Sys.time()
# names(result_fgsea) <- colnames(analysis_2_t_argu[, 1:n])
names(result_fgsea) <- colnames(analysis_2_t_argu)
gc(T,T,T)
gc(T,T,T)fgseaの結果はリストで返ってくる。必要な統計値等をそのリストから出してきて、それぞれをデータフレームとして保存する。ここでは、Enrichment Score(ES)、Normalized Enrichment Score(NES)、padj(FDR補正後のP値)をそれぞれデータフレームresult_fgsea_ES、result_fgsea_NES、result_fgsea_padjとして作成する。
# code 105
# Extract results of fgsea, foreach, hallmark
result_fgsea_ES <- data.frame()
result_fgsea_NES <- data.frame()
result_fgsea_padj <- data.frame()
for(i in 1:length(result_fgsea)){
result_fgsea_ES[1:nrow(result_fgsea[[i]]),i] <- data.frame(result_fgsea[[i]]$ES)
result_fgsea_NES[1:nrow(result_fgsea[[i]]),i] <- data.frame(result_fgsea[[i]]$NES)
result_fgsea_padj[1:nrow(result_fgsea[[i]]),i] <- data.frame(result_fgsea[[i]]$padj)
}
colnames(result_fgsea_ES) <- colnames(analysis_2_t_argu)
rownames(result_fgsea_ES) <- result_fgsea[[1]]$pathway # any component can be used for rownames() because gene set name in all components is same.
colnames(result_fgsea_NES) <- colnames(analysis_2_t_argu)
rownames(result_fgsea_NES) <- result_fgsea[[1]]$pathway # any component can be used for rownames() because gene set name in all components is same.
colnames(result_fgsea_padj) <- colnames(analysis_2_t_argu)
rownames(result_fgsea_padj) <- result_fgsea[[1]]$pathway # any component can be used for rownames() because gene set name in all components is same.
gc(T,T,T)
gc(T,T,T)code 106は各エンリッチメント解析に要した時間を取得する。5200タンパク質くらいなので、1時間ちょっとで終了出来る。
# code 106
end_results_escape - start_results_escape # Time difference of 5.852598 mins
end_results_GSVA- start_results_GSVA # Time difference of 4.735823 mins
end_results_fgsea - start_results_fgsea # Time difference of 1.280739 hours言うても下手すると2時間以上は掛かるので、ここでRDataとして保存しておく。
# code 107
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_5.RData")escape、gsva、fgseaによるエンリッチメント解析の結果をまとめる
escapeによって得られた結果をデータフレームresult_escape_dfに入れる。列genesetを作成し、そこに各遺伝子セット名とパッケージ名(6行目で列名をクセで列libraryとして作成してしまった)とアンダーバーで繋いでおく。こうしなければ、後のgsvaやfgseaの結果と区別がつかなくなってしまう。そしてこのデータフレームresult_escape_dfでも列ID_2を行名にしておく。
# code 108
# escape
column_escape <- colnames(result_escape) %>% data.frame()
colnames(column_escape) = "geneset"
column_escape <- column_escape %>% mutate(library = "escape")
column_escape <- column_escape %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_escape) <- column_escape$geneset
result_escape_df <- as.data.frame(result_escape) %>% rownames_to_column(var = "ID_2")続いてescapeの場合と同様にgsvaの結果を処理する。gsvaの結果は列が遺伝子セット名、行がタンパク質発現プロファイル名となる。これはescapeの結果に合わせて、t()で転置させておき、順次、escapeの場合と同様の行列のフォーマットにし、同様の処理を行っていき、データフレームresult_gsva_dfを作成する。
# code 109
# gsva
## prepare dataset
result_gsva_t <- result_gsva %>% t() %>% data.frame()
## prepare column name, hallmark
column_gsva <- colnames(result_gsva_t) %>% data.frame()
colnames(column_gsva) = "geneset"
column_gsva <- column_gsva %>% mutate(library = "gsva")
column_gsva <- column_gsva %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_gsva_t) <- column_gsva$geneset
result_gsva_df <- result_gsva_t %>% rownames_to_column(var = "ID_2")gseaの結果は複数あるが、ここでマトリックスresult_fgsea_NESを使用する。これもgsvaと同様にt()で転置させて行を遺伝子セット名、列をタンパク質発現プロファイル名にし、上記のescapeとgvsaと同じような行列のフォーマットでデータフレームresult_fgsea_dfを作成する。
# code 110
# fgsea
result_fgsea_t <- t(result_fgsea_NES) %>% data.frame()
## prepare column name, hallmark
column_fgsea <- colnames(result_fgsea_t) %>% data.frame()
colnames(column_fgsea) = "geneset"
column_fgsea <- column_fgsea %>% mutate(library = "fgsea")
column_fgsea <- column_fgsea %>% unite(geneset, library, sep = "_", col = "geneset", remove = TRUE)
colnames(result_fgsea_t) <- column_fgsea$geneset
result_fgsea_df <- result_fgsea_t %>% rownames_to_column(var = "ID_2")
各エンリッチメント解析の結果をデータフレームに変換したら、それらをリストtempに入れていき、1つに結合する。エンリッチメント解析に使用した元のデータも必要、というか、どちらかと言ったら元のデータの方が重要なので、データフレームanalysis(これは、タンパク質発現量をその中央値で正規化した底2の対数)もリストtempに入れる。それをreduce()と使って各リストにinner_join()を適用していく。そのときreduce()を使う場合はinner_join()の引数を設定することが出来ないので、必要な引数を予め設定しておいた関数inner_join_ID_2を定義しておき、それをreduce()でリストの構成要素の適用する。
# code 111
# merge all dataset
# analysis_2_t_argu
view(analysis_2_t_argu[1:100, 1:100])
view(analysis[1:100, 1:100])
view(result_escape_df[1:100, 1:100])
view(result_gsva_df[1:100, 1:100])
view(result_fgsea_df[1:100, 1:100])
# analysis_2_argu_df <- analysis_2_t_argu_df %>% column_to_rownames(var = "protein") %>% t() %>% data.frame() %>% rownames_to_column(var = "ID_2")
temp <- list()
temp[[1]] <- analysis
temp[[2]] <- result_escape_df
temp[[3]] <- result_gsva_df
temp[[4]] <- result_fgsea_df
inner_join_ID_2 <- function(x, y){inner_join(x, y, by = "ID_2")}
all_1 <- reduce(temp, inner_join_ID_2) %>% data.frame()
# column 269; ID_2
# column 270 - 5477; protein expression.
# column 5530 - ncol(analysis); results of enrichment analysis.
# other columns; clinical information.
all_1 <- all_1 %>% dplyr::select(ID_2, c(270:5477, 5478:ncol(all_1), 1:268))
# column 2 - 5209; protein expression.
# column 5210 - 81851; results of enrichment analysis.
# other columns; clinical information. reduce()には結構時間が掛かるので、それが終わったらRDataとして外部に保存しておく。どうやら、処理自体はそこそこ早く終わるもののデータの移動に時間が掛かっている印象を受ける。
# code 112
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_5.RData")疾患名の整理
ここから、疾患を選んでいく。疾患選びに使える列は、列Disease_Typeや列Tissue_Typeである。列Disease_Typeには疾患(がんや腫瘍)の種類が、列Tissue_Typeには正常組織かがん組織かが入力してある。列Disease_Type(元々データフレームbiospeciemenに入っていた列)、列Disease_Type_clinical(元々データフレームclinicalに入っていた列)、列Tissue_Type(元々データフレームbiospeciemenに入っていた列)のクロス集計をtable()で出し、結果をデータフレームdiseaseに入れ、頻度を見ながら解析に使える疾患を選ぶ。このバージョンのCPTACには列Disease_Typeの値「Meningiomas」が1例しか登録されていなかった。1例だけだと解析にはならないので、このような疾患は後ほど除く。次に列Disease_Type_Abbreviation、列Tissue_Type_2を新しく作り、それに、それぞれ疾患の略称、空欄をアンダーバーで置き換えた列Tissue_Typeの値を入れ、その2つの列の値をアンダーバーで繋いだ値を列Tissue_Diseaseを作成し、データフレームdisease_2を作成する。この略称は、TCGAっぽくしているが、短くて判別できるならば何でも良いと思う。そこから必要な列を選んでデータフレームdisease_3を作成し、列Disease_Typeと列Tissue_Typeの値で並べ替えておく。データフレームall_2とデータフレームdisease_3を列Disease_Typeと列Tissue_Typeの値でinner_join()し、データフレームall_1にデータフレームdisease_3の列を加えたデータフレームall_2を作成する。そこから、列Disease_Typeの値が「Meningiomas」の例を除く。
# code 113
# There is probably no problem
table(all_1$Project_Name) %>% data.frame() %>% view()
table(all_1$Program_Name) %>% data.frame() %>% view()
table(all_1$Disease_Type, all_1$Tissue_Type) %>% data.frame() %>% view()
table(all_1$Disease_Type)
table(all_1$Disease_Type_clinical)
table(all_1$Tissue_Type)
#
disease <- table(all_1$Disease_Type,
all_1$Disease_Type_clinical,
all_1$Tissue_Type) %>% data.frame() %>% rename("Disease_Type" = "Var1",
"Disease_Type_clinical" = "Var2",
"Tissue_Type" = "Var3")
table(disease$Disease_Type) %>% data.frame() %>% nrow() # 17
table(disease$Disease_Type %in% disease$Disease_Type_clinical)
# TRUE
# 867
table(disease$Disease_Type) %>% data.frame() %>% filter(Freq >= 1) %>% view()# Disease_Type == "Meningiomas" is just 1 case and therefore Freq should be >= 1.
disease <- disease %>% filter(Freq >= 1)
table(disease$Disease_Type %in% disease$Disease_Type_clinical)
# TRUE
# 28
table(disease$Disease_Type)
# https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations
disease[disease$Disease_Type == "Meningiomas",] %>% view() # "Meningiomas" is just 1 case.
#
disease_2 <- disease %>% mutate(
Disease_Type_Abbreviation = case_when(Disease_Type == "Breast Invasive Carcinoma"~ "BRCA",
Disease_Type == "Cholangiocarcinoma"~ "Cholangiocarcinoma",
Disease_Type == "Clear Cell Renal Cell Carcinoma"~ "ccRCC",
Disease_Type == "Early Onset Gastric Cancer"~ "Gastric",
Disease_Type == "Glioblastoma"~ "GBM",
Disease_Type == "Gliomas"~ "Gliomas",
Disease_Type == "Head and Neck Squamous Cell Carcinoma"~ "HNSCC",
Disease_Type == "Hepatocellular Carcinoma"~ "LIHC",
Disease_Type == "Lung Adenocarcinoma"~ "LUAD",
Disease_Type == "Lung Squamous Cell Carcinoma"~ "LUSC",
Disease_Type == "Meningiomas" ~ "Meningiomas",
Disease_Type == "Non-Clear Cell Renal Cell Carcinoma"~ "non-ccRCC",
Disease_Type == "Other"~ "Other",
Disease_Type == "Ovarian Serous Cystadenocarcinoma"~ "OV",
Disease_Type == "Pancreatic Ductal Adenocarcinoma"~ "PAAD",
Disease_Type == "Pediatric/AYA Brain Tumors"~ "Peds_AYA_Brain",
Disease_Type == "Uterine Corpus Endometrial Carcinoma"~ "UCEC",
Disease_Type == TRUE ~ NA),
Tissue_Type_2 = case_when(Tissue_Type == "Normal"~ "Normal",
Tissue_Type == "Tumor"~ "Tumor",
Tissue_Type == "Not Reported"~ "Not_Reported",
Tissue_Type == TRUE ~ NA)
) %>% unite(col = Tissue_Disease, c("Tissue_Type_2", "Disease_Type_Abbreviation"), sep = "_", remove = FALSE)
disease_3 <- disease_2 %>% select("Disease_Type", "Tissue_Type", "Disease_Type_Abbreviation", "Tissue_Disease")
disease_3 <- disease_3 %>% arrange(Disease_Type, Tissue_Type)
all_2 <- left_join(all_1, disease_3, by = c("Tissue_Type", "Disease_Type"))
check_disease_matching <- table(all_2$Disease_Type,
all_2$Disease_Type_clinical,
all_2$Tissue_Type,
all_2$Tissue_Disease) %>% data.frame() %>% rename("Disease_Type" = "Var1",
"Disease_Type_clinical" = "Var2",
"Tissue_Type" = "Var3",
"Tissue_Disease" = "Var4") %>% filter(Freq >=1)
all_2[all_2$ID_2 == "case4602",]$Disease_Type # Meningiomas
all_2[all_2$ID_2 == "case4602",]$Tissue_Type # Tumor
列Tissue_Typeには、値が「Not Reported」という謎症例が登録されている。絶対に何かの試料なわけだから、それをちゃんと書いてから登録してほしいものだ。これ自分で解析していて困らないのだろうか…列Tissue_Typeの値が「Not Reported」の例は、正直どのように解析したら良いかわからないので、この症例は除き、データフレームall_3を作成する。
ここで疾患が何とかなったので、症例数が少なすぎる疾患は除く。ここでは登録された症例数が10より少ない疾患は除く。table()で関心のある列のクロス集計表をだし、それをデータフレームcheck_disease_matching_all_3にして、その列Freqが10より少ないものを抽出して、データフレームcheck_disease_matching_all_3_to_be_removedを作成する。このとき、列Disease_Typeと列Disease_Type_clinicalが一致しているか、列Tissue_Typeと列Tissue_Diseaseにおかしな不一致はないかも同時に確認する。データフレームcheck_disease_matching_all_3_to_be_removedを使って、データフレームall_3から症例数が10より少ない疾患を除き、それをデータフレームall_4とする。
# code 114
disease_4 <- disease_3 %>% filter(Tissue_Type != "Not Reported")
all_3 <- all_2 %>% filter(Tissue_Type != "Not Reported")
check_disease_matching_all_3 <- table(all_3$Disease_Type,
all_3$Disease_Type_clinical,
all_3$Tissue_Type,
all_3$Tissue_Disease) %>% data.frame() %>% rename("Disease_Type" = "Var1",
"Disease_Type_clinical" = "Var2",
"Tissue_Type" = "Var3",
"Tissue_Disease" = "Var4") %>% filter(Freq >= 1)
check_disease_matching_all_3_to_be_removed <- check_disease_matching %>% dplyr::filter(Freq < 10)
all_4 <- all_3[all_3$Tissue_Disease %in% check_disease_matching_all_3_to_be_removed$Tissue_Disease == FALSE, ]ここでは、のちのちバイオリンプロットやカプランマイヤー生存曲線で表示するための順番を設定するために使用するfactor()のために、疾患の順番を決める。上記で行ってきた方法と同様に、データフレームall_4からtable()で列Disease_Type、列Disease_Type_clinical、列Tissue_Type、列Tissue_Diseaseの集計表を求め、それをデータフレームdisease_5とし、それを列Disease_Type、列Tissue_Typeの値で並び替えてデータフレームdisease_6とする。このデータフレームdisease_6の列Tissue_Diseaseの値をベクトルorder_diseaseとして保持しておき、必要になったらデータフレームのfactor()に使用する。今後、何らかの疾患を除く可能性もあるので、まだfactor()は設定しないでおく(factor()を設定した上で、何らかの疾患が除かれた場合、グラフにその疾患が0として表示されてしまうため)。
# code 115
# for setting factor
disease_5 <- table(all_4$Disease_Type,
all_4$Disease_Type_clinical,
all_4$Tissue_Type,
all_4$Tissue_Disease) %>% data.frame() %>% rename("Disease_Type" = "Var1",
"Disease_Type_clinical" = "Var2",
"Tissue_Type" = "Var3",
"Tissue_Disease" = "Var4") %>% filter(Freq >= 1)
table(disease_5$Disease_Type %in% disease_5$Disease_Type_clinical)
# TRUE
# 24
disease_6 <- disease_5 %>% arrange(Disease_Type, Tissue_Type)
# disease_7 <- left_join(disease_6, disease_4, by = c("Disease_Type", "Tissue_Type")) #%>% select("Disease_Type", "Tissue_Type", "Tissue_Disease")
table(disease_6$Disease_Type %in% disease_6$Disease_Type_clinical)
# TRUE
# 24
check_Tissue_Disease <- table(all_4$Tissue_Disease) %>% data.frame() %>% rename("Tissue_Disease" = "Var1")
table(check_Tissue_Disease$Tissue_Disease) %>% data.frame() %>% rename("Tissue_Disease" = "Var1") %>% view()
table(all_4$Tissue_Disease %in% disease_6$Tissue_Disease)
# TRUE
# 2465
order_disease <- disease_6$Tissue_Disease
length(order_disease) # 24
table(all_4$Tissue_Disease %in% order_disease)
# TRUE
# 2465
# This is not yet. it needs to be done just before violin plot. Move to just before violin plot, around line 1028
# all_4$Tissue_Disease <- factor(all_4$Tissue_Disease,
# levels = order_disease,
# labels = order_disease,
# exclude = NA, ordered = is.ordered(all_4$Tissue_Disease), nmax = NA)タンパク質発現量やエンリッチメントスコアのヒストグラムを表示してみる
大体、解析に使用するデータフレームが整ったので、ここで関心のあるタンパク質もしくは遺伝子セットのエンリッチメントスコアのヒストグラムと四分位数も出してみる。言うて、これらを出したところで何もわからないが。以下はGSVAで求めたHALLMARK_INTERFERON_GAMMA_RESPONSEのエンリッチメントスコアに関する値である。
# code 116
# histogram
# gvsa
#geom_histogram(binwidth = 10*(1/nrow(all_4)))
ggplot(all_4, aes(x = HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva)) +
geom_histogram() +
xlim(range(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva))
median(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva) # 0.04983395
range(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva) # -0.6655193 0.6942024
quantile(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva, probs = c(0.25, 0.5, 0.75)) # -0.28484209 0.04983395 0.32138058以下はescapeで求めたHALLMARK_INTERFERON_GAMMA_RESPONSEのエンリッチメントスコアに関する値である。
# code 117
# histogram
# escape
#binwidth = 10*(1/nrow(all_4))
ggplot(all_4, aes(x = HALLMARK_INTERFERON_GAMMA_RESPONSE_escape)) +
geom_histogram() +
xlim(range(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape))
#
median(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape) # 193.8187
range(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape) # -1466.005 1900.957
quantile(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape, probs = c(0.25, 0.5, 0.75)) # -349.2947 193.8187 737.5548
min(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape) # -1466.005
max(all_4$HALLMARK_INTERFERON_GAMMA_RESPONSE_escape) # 1900.957ここで、疾患ごとの症例数を、列Tissue_Diseaseの値に付ける。まず、データフレームall_4を列Tissue_Diseaseでgroup_by()に入れる。こうすることで、列Tissue_Diseaseの値毎に処理することが出来る。今回は症例に通し番号を付ける。列Tissue_Diseaseで疾患毎にグループ化し、そのグループ内にある症例数をn()で求め、1からn()の値を症例に振っていく。それをmutate()で列case_numberとして入れ、データフレームall_5とする。そして列Tissue_Diseaseと作成した列case_numberをunite()で繋いで、列Tissue_Disease_caseを作成する。これでTumor_LUAD1からNormal_LUAD101のように、LUADの例が101例あるならば、1から順番に101まで番号を振る。ここでは正常とがん組織を区別しなかったので、Tumor_LUAD1からNormal_LUAD101のような値になっている。これだとがんのみ抽出したときに連番にならないが、それでも個別に識別できるし、降順もしくは昇順に並べることが出来るので問題ない。
# code 118
# required data; all_4
# n() returns current group size.
all_5 <- all_4 %>% group_by(Tissue_Disease) %>% mutate(
case_number = rep(1:n(), each = 1)
)
all_5 <- all_5 %>% unite(col = "Tissue_Disease_case", c("Tissue_Disease", "case_number"), sep = "", remove = FALSE)バイオリンプロットで疾患ごとのタンパク質発現量を比較する
ここから、あるタンパク質の発現量が疾患毎にどのくらい違うのか見ていく。単にバイオリンプロットを描くだけでなく、そのプロット上に全症例のの25パーセンタイル、中央値、75パーセンタイルを示す水平線を引き、関心のある疾患では、全体に比べてどのくらいの発現量を持つのかわかるようにする。また、その発現量に応じてバイオリンプロットの色を変えて、見分けがつくようにする。
バイオリンプロットのためのデータを準備する
データフレームall_5から、必要な列を抽出してくる。各タンパク質名と遺伝子セット名を1つずつ指定するには大量過ぎるので、ここでは以下の表の通り、列番号で抽出する。
| 使用するデータフレーム | 列番号 | 列名 | 値の例 |
|---|---|---|---|
| all_5 | 2 – 5209 | 各タンパク質 | 発現量 |
| all_5 | 5210 – 81851 | 各遺伝子セット | エンリッチメントスコア |
| all_5 | 82121 | Tissue_Disease_case | Tumor_LUAD1 … |
| all_5 | 82120 | Disease_Type_Abbreviation | LUAD … |
| all_5 | 82122 | Tissue_Disease | Tumor_LUAD … |
| all_5 | 81897 | Tissue_type | Tumor |
まず、データフレームall_5から列Tissue_typeが「Tumor」の症例をそれぞれデータフレームを選んできて、データフレームall_5_tumorとする。これは、バイオリンプロットに表示する中央値などの計算はがん組織のみの値で計算するためである。疾患を標的にする場合、正常組織とがん組織を含めたタンパク質発現量の中央値は意味がない。正常組織とがん組織の比較は、後にWilcoxonの中央値検定で比較する。
がん組織における発現量をタンパク質もしくは遺伝子セット毎に求めるためには、各タンパク質もしくは各遺伝セットの上記の表の値を、リストの要素に1つずつ入れていくのが良いと思う。今回はリストproteinとリストenrichmentの各要素に入れる。入れたら2つのリストをc()で縦方向に結合させ、リストstatsを作成する。正直、これはタンパク質と遺伝子セットを別々の処理で読んでくる必要はないと思う。要はリストstatsに最初からall_5_tumor[,2:81851]を列毎に読んでくれば良いと思う。
そして、リストstatsの各要素をデータフレームに変換し、それをリストstats_2の各要素に入れていく。今後のためもリストの要素にはnames()で名前を付けておく必要がある。これはリストが出来るたびに行っている。このリストstats_2は、各要素毎の中央値などを計算するために使用する。
次に、同様の方法で、今度は正常組織も含めたリストstats_w_normal_2を作成する。これは、バイオリンプロットの表示に使用する。
# code 119
# violin plot
## prepare data
### tumor only
#### each gene; 2 - 5209
##### Tumor_LUAD1 ... ; Tissue_Disease_case; 82121
##### LUAD ...; Disease_Type_Abbreviation ; 82120
##### Tumor_LUAD ... ; Tissue_Disease; 82122
##### Tumor, Normal ; Tissue_type; 81897
which(colnames(all_5) == "Tissue_Disease_case") # 82121 ;Tumor_LUAD1
which(colnames(all_5) == "Disease_Type_Abbreviation") # 82120; LUAD
which(colnames(all_5) == "Tissue_Disease") # 82122; Tumor_LUAD
which(colnames(all_5) == "Tissue_Type") # 81897; Tumor
# all_5[,2:5209] <- apply(all_5[,2:5209], 2, function(x) 2^x) %>% data.frame()
# all_5[,2:81851] <- apply(all_5[,2:81851], 2, function(x) as.numeric(x)) %>% data.frame()
# Prepare dataset for quantile of tumor
## The quantiles of TUMOR, but not NORMAL, were required. Therefore, the dataset was filtered for TUMOR only.
all_5_tumor <- all_5 %>% filter(Tissue_Type == "Tumor")
### each protein.
protein <- list()
for(i in 1:ncol(all_5_tumor[,2:5209])){
protein[[i]] <- all_5_tumor[,c(1, 82121, 82120, 82122, 81897, 2+i-1)]
}
names(protein) <- colnames(all_5[,2:5209])
#### each enrichment.
enrichment <- list()
for(i in 1:ncol(all_5_tumor[,5210:81851])){
enrichment[[i]] <- all_5_tumor[,c(1, 82121, 82120, 82122, 81897, 5210+i-1)]
}
names(enrichment) <- colnames(all_5_tumor[,5210:81851])
# combine list "protein" and list "enrichment."
stats <- c(protein, enrichment)
# each component in list "stats" must be data frame for calculation statistical values.
stats_2 <- list()
for(i in 1:length(stats)){
stats_2[[i]] <- data.frame(stats[[i]])
}
names(stats_2) <- names(stats)
stats_2[[5210]] %>% view()
### dataset with normal
#### each gene
protein_w_normal <- list()
for(i in 1:ncol(all_5[,2:5209])){
protein_w_normal[[i]] <- all_5[,c(1, 82121, 82120, 82122, 81897, 2+i-1)]
}
names(protein_w_normal) <- colnames(all_5[,2:5209])
enrichment_w_normal <- list()
#### each geneset
for(i in 1:ncol(all_5[,5210:81851])){
enrichment_w_normal[[i]] <- all_5[,c(1, 82121, 82120, 82122, 81897, 5210+i-1)]
}
names(enrichment_w_normal) <- colnames(all_5[,5210:81851])
stats_w_normal <- c(protein_w_normal, enrichment_w_normal)
# each component in list "stats" must be data frame for calculation statistical values.
stats_w_normal_2 <- list()
for(i in 1:length(stats)){
stats_w_normal_2[[i]] <- data.frame(stats_w_normal[[i]])
}
names(stats_w_normal_2) <- names(stats_w_normal)バイオリンプロットのための四分位数を計算する
上記で作成したリストstats_2の各要素毎に、四分位数をquantile()で計算していき、リストstats_2の各構成要素の中央値、25パーセンタイル、75パーセンタイルをそれぞれリストQ50_all、Q25_all、Q75_allに入れていく。リストにはタンパク質名と遺伝子セット名をnames()で入れる。
# code 120
#
#
#
#### stats for all disease
which(is.na(all_5), arr.ind = TRUE) # There are several NA in all_5.
Q50_all <- list()
Q25_all <- list()
Q75_all <- list()
for(i in 1:length(stats_2)){
print(paste0("### Now ", i, " out of ", length(stats_2), " is processing. ###"))
Q50_all[i] <- median(stats_2[[i]][,6], na.rm = TRUE)
Q25_all[[i]] <- quantile(stats_2[[i]][,6], probs = 0.25, na.rm = TRUE)
Q75_all[[i]] <- quantile(stats_2[[i]][,6], probs = 0.75, na.rm = TRUE)
}
names(Q50_all) <- names(stats_2)
names(Q25_all) <- names(stats_2)
names(Q75_all) <- names(stats_2)
上記のcode 120でタンパク質もしくは遺伝子セット毎の疾患全体の四分位数を求めたが、code 121では、タンパク質もしくは遺伝子セットの四分位数を疾患毎にを求めていく。ここでは、四分位数だでなく、平均値、標準偏差、最大値、最小値、疾患の症例数、疾患全体に対する対象となる疾患のタンパク質発現量もしくはエンリッチメントスコアの中央値の比を求め、それらをリストstats_3の各要素に入れていく。、ここで求めた疾患のごと中央値のと、疾患全体の中央値を比較して、特にその中央値が疾患の中でも高いのか低いのかをバイオリンプロット上に表示する。以下の表が列名と値である。
| 作成する列 | 値 |
|---|---|
| mean | その疾患における平均値 |
| sd_each | その疾患における標準偏差 |
| Q50_each | その疾患における中央値 |
| Q25_each | その疾患における25パーセンタイル |
| Q75_each | その疾患における75パーセンタイル |
| min_each | その疾患における最小値 |
| max_each | その疾患における最大値 |
| FC_each | その疾患における中央値/疾患全体における中央値 |
| n_each | その疾患における症例数 |
そして、特に四分位数の値を階層化し、その階層をバイオリンプリプトットの色に使用する。
| 列 | 値の範囲 | 列colorの値 |
|---|---|---|
| color | 各疾患の中央値(列Q50_each)が疾患全体の25パーセンタイル(列Q25_all)以下 | 0_low |
| color | 各疾患の中央値(列Q50_each)が疾患全体の25パーセンタイル(列Q25_all)以上 | 2_high |
| color | 各疾患の中央値(列Q50_each)が疾患全体の25パーセンタイル(列Q25_all)より多く、25パーセンタイル(列Q25_all)より低い | 1_even |
code 121では、個人的にはデータの解析ではあまり馴染みの無い方法を使う。code 121の12行目にデータフレームとして.dataを指定しているが、12行目に指定したデータフレームdfを動的に参照する方法である。個人的にはパイプ(%>%)で繋いでいるときにのみ有効であり、今回のようにfor()の各ステップ毎にdfの内容が変わっていくようなときは便利である。
# code 121
#### stats for each signature
stats_3 <- list()
for (i in 1:length(stats_2)) {
print(paste0("### Now ", i, " out of ", length(stats_2), " is processing. ###"))
df <- stats_2[[i]]
geneset <- names(df)[6]
##### .data[[geneset]] is dynamic reference in data.frame and tibble, not list.
stats_3[[i]] <- df %>% group_by(Disease_Type_Abbreviation) %>%
summarise(
mean = mean(.data[[geneset]], na.rm = TRUE),
sd_each = sd(.data[[geneset]], na.rm = TRUE),
Q50_each = quantile(.data[[geneset]], probs = 0.5, na.rm = TRUE),
Q25_each = quantile(.data[[geneset]], probs = 0.25, na.rm = TRUE),
Q75_each = quantile(.data[[geneset]], probs = 0.75, na.rm = TRUE),
min_each = min(.data[[geneset]], na.rm = TRUE),
max_each = max(.data[[geneset]], na.rm = TRUE),
FC_each = quantile(.data[[geneset]], probs = 0.5, na.rm = TRUE) / Q50_all[[i]],
n_each = n(),
.groups = "drop"
) %>% mutate(
color = case_when(
Q50_each <= Q25_all[[i]] ~ "0_low",
Q50_each >= Q75_all[[i]] ~ "2_high",
Q50_each > Q25_all[[i]] & Q50_each < Q75_all[[i]] ~ "1_even"))
}
names(stats_3) <- names(stats)
view(stats_3[[1]])
view(stats_2[[1]])
view(stats_w_normal[[1]])code 121の計算はがん組織のみの統計量である。これを正常組織も含んだデータであるリストstats_w_normal_2の要素にも反映していく。このために、match()を使用する。これは%in%と同じであるので、それで短く書き換えられるような気がするが、なんか複雑になりそうなのでmatch()を使った。
code 121の9行目で、リストstats_w_normalの要素の1つ目に入っているデータフレームA1BGの列Disease_Type_Abbreviationの値「LUAD」は、リストstats_3の要素1つ目にあるデータフレームA1BGの列Disease_Type_Abbreviationの値のどの場所(つまり何行目か)と一致するのかを、match()で取得し、その場所(行)をリストcolor_matchingの1つ目の構成要素color_matching[[1]]に入れる。この結果、color_matching[[1]]には、リストstats_w_normalの1つ目の要素に入ってるデータフレームA1BG(stats_w_normal[[1]])の列Disease_Type_Abbreviationの値は、リストstats_3の1つ目の要素(stats_3[[1]]、データフレームの名前はA1BG)の何行目にあるのか、という値がベクトルとして入る。そして10行目では、リストstats_3の要素1つ目に入っているデータフレームA1BGの列colorの値のうち、color_matching[[1]]で示される行にある値をリストstats_w_normal_2の要素1つ目のデータフレームA1BGの列colorに入れる。こうして、code 121で作成したがん組織のタンパク質発現量もしくはエンリッチメントスコアの中央値による階層(リストstats_3の各要素の列colorの値)を、リストstats_w_normalの各要素のデータフレームに列colorとして入れていく。これで、リストstats_w_normalの各要素にも階層化、すなわちバイオリンプロットの色を付けることが出来る。
このcode 121の最後に、バイオリンプロットで表示するタンパク質もしくは遺伝子セットを選ぶためのデータフレームprotein_geneset_nameを作成しておく。これは単にタンパク質もしくは遺伝子セット名だけからなるデータフレームである。
# code 122
# #### color matching
length(stats_w_normal_2[[1]]$Disease_Type_Abbreviation) # 2465
length(stats_3[[1]]$Disease_Type_Abbreviation) # 14
color_matching <- list()
for (i in 1:length(stats_2)) {
color_matching[[i]] <- match(stats_w_normal_2[[i]]$Disease_Type_Abbreviation, stats_3[[i]]$Disease_Type_Abbreviation)
stats_w_normal_2[[i]]$color <- stats_3[[i]]$color[color_matching[[i]]]
}
table(stats_w_normal_2[[3]]$color) # Check the code was running normally
#### If log10 scale is better for y-axis, use "scale_y_log10() +" before "theme()"
#### Check the gene or geneset name you need.
protein_geneset_name <- data.frame(target_protein_geneset = names(stats_3)) 正常組織と腫瘍(がん)組織間でWilcoxonの中央値検定を行う
次に、正常組織とがん組織のタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアを、各疾患毎にウィルコクソンの中央値検定(wilcox.test())を使って検定する。
まず、7行目で疾患名を得るためにリストstats_3の1つ目の構成要素であるデータフレームA1BG(これは1つ目でなくても良い。全部同じはずなので、どこでも良い)の列Disease_Type_Abbreviationから、疾患名を入れたデータフレームdiseaseを作成する。次に、12、13行目で、リストwilcoxとtempを用意する。
37行目からがwilcox.testである。リストstats_w_normal_2の各要素に入っているデータフレームを、列Tissue_Typeの値でpivot_wider()を行うことで発現量を列Tumorと列Normalに分け、その2つの列、列Tumorと列Normal間でwilcox.test()を行い、その結果得られたp値を列pvalueに入れていく。しかしながら、どうしても列Normalの値がない症例もある。その場合はwilcox.test()はエラーになってしまうので、tryCatch()を使ってもしエラーが出たらそのときの列pvalueの値に「NA」を入れていく。さらに、ここで得られた列pvalueの値を使って、以下の表の様に階層化していく。
| 条件1 | 理論演算子 | 条件2 | 作成する列 | 入れる値 |
|---|---|---|---|---|
| 列Tissue_Diseaseに文字列「Tumor_」を含む | & | 列colorが「0_low」 | color_2 | 0_low |
| 列Tissue_Diseaseに文字列「Tumor_」を含む | & | 列colorが「1_even」 | color_2 | 1_even |
| 列Tissue_Diseaseに文字列「Tumor_」を含む | & | 列colorが「2_high」 | color_2 | 2_high |
| 列Tissue_Diseaseに文字列「Tumor_」を含む | & | 列significanceが「3_significant」 | color_2 | 3_significant |
| 列Tissue_Diseaseに文字列「Normal_」を含む | & | 列significanceが「3_significant」 | color_2 | 3_significant |
| 列Tissue_Diseaseに文字列「Normal_」を含む | & | 列significanceが「4_not_significant」 | color_2 | 4_not_significant |
| 列Tissue_Diseaseに文字列「Normal_」を含む | & | 列significanceがNA | color_2 | 4_not_significant |
| 他 | – | – | color_2 | NA |
そして、必要な列ID_2、列Tissue_Disease_case、列Disease_Type_Abbreviation、列Tissue_Disease、列color、列color_2、列p_value、列significanceを選んできて、リストtempの各要素に入れる。リストtempは14個の要素、すなわち疾患毎になっているはずである。それをリストwilcoxの要素として順次入れていく。リストwilcoxの要素は各タンパク質もしくは各遺伝子セットになっているはずである。つまりリストwilcoxには各タンパク質もしくは各遺伝子セット毎に構成され、その中にさらに各疾患ごとの検定や階層の値がネストして入っていることになる。
がん組織のプロファイルは疾患全体と比較することで既に階層化されいる(すなわち腫瘍組織は既に色が割り当てられている)ので、wilcox.test()による結果は正常組織側に反映する(すなわち正常組織とがん組織のタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアに統計的有意差が検出されたら、正常組織側の色を変える)ことにする。
# code 123
# wilcox.test, normal vs tumor
view(stats_3[[1]])
disease <- table(stats_3[[1]]$Disease_Type_Abbreviation) %>% data.frame()
disease <- disease %>% rename("Disease_Type_Abbreviation" = "Var1")
disease[1,1]
wilcox <- list()
temp <- list()
start_wilcox <- Sys.time()
## Following is for checking the code.
# for(x in 1:5){
# print(paste0("Now processing ", x, " out of ",length(stats_3)," genes"))
# for(y in 1:nrow(disease)){
# temp[[y]] <- stats_w_normal[[x]][stats_w_normal[[x]]$Disease_Type_Abbreviation %in% disease[y,1],] %>% pivot_wider(
# names_from = Tissue_Type,
# values_from = names(stats_w_normal)[x]) %>% mutate(
# p_value = tryCatch({wilcox.test(Tumor, Normal, alternative = "two.sided")$p.value}, error = function(e) {NA})
# )
# }
# }
#
check <- names(stats_w_normal_2) %>% data.frame() %>% rename("protein_geneset" = ".")
stats_w_normal_2[["chr10p12_escape"]] %>% view()
names(stats_w_normal_2[[5210]])
# for(x in 1:200){
# for(x in 1:length(stats_w_normal)){
# for(x in 1:length(stats_3)){
for(x in 1:length(stats_3)){
print(paste0("Now processing ", x, " out of ",length(stats_3)," genes"))
for(y in 1:nrow(disease)){
temp[[y]] <- stats_w_normal_2[[x]][stats_w_normal_2[[x]]$Disease_Type_Abbreviation %in% disease[y,1],] %>% pivot_wider(
names_from = Tissue_Type,
values_from = names(stats_w_normal_2)[x]) %>% mutate(
p_value = tryCatch({wilcox.test(Tumor, Normal, alternative = "two.sided")$p.value}, error = function(e) {NA})
) %>% mutate(
significance = case_when(
p_value < 0.05 ~ "3_significant",
p_value >= 0.05 ~ "4_not_significant",
TRUE ~ NA
)
) %>% mutate(
color_2 =case_when(
str_detect(Tissue_Disease, "Tumor_") == TRUE & color == "0_low" ~ "0_low",
str_detect(Tissue_Disease, "Tumor_") == TRUE & color == "1_even" ~ "1_even",
str_detect(Tissue_Disease, "Tumor_") == TRUE & color == "2_high" ~ "2_high",
str_detect(Tissue_Disease, "Normal_") == TRUE & significance == "3_significant" ~ "3_significant",
str_detect(Tissue_Disease, "Normal_") == TRUE & significance == "4_not_significant" ~ "4_not_significant",
str_detect(Tissue_Disease, "Normal_") == TRUE & significance == NA ~ "4_not_significant",
TRUE ~ NA
)
) %>% select(ID_2, Tissue_Disease_case, Disease_Type_Abbreviation, Tissue_Disease, color, color_2, p_value,significance)
}
names(temp) <- disease$Disease_Type_Abbreviation
wilcox[[x]] <- temp
}
# names(wilcox) <- names(stats_3)[1:200]
names(wilcox) <- names(stats_3)
end_wilcox <- Sys.time()
# at the end of the for() chunk, pivot_longer will be better than select() but several dataset does not have Normal column and therefore pivot_longer() produces error.
duration_wilcox <- end_wilcox - start_wilcox # about 8 hours spent.ここまでの結果をRDataとして外部に保存する。処理にかなりの時間を要するためである。
# code 124
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_6.RData")作成したリストwilcoxの中身である各疾患ごとの階層化と有意差をrbind()で縦方向(行方向)に結合し、リストwilcox_merge_1の各要素に入れていく。それをリストstats_w_normal_2の各要素とinner_join()し、リストstats_w_normal_w_wilcoxを作成する。これでタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアの階層化が終了である。この階層化は、バイオリンプロットの疾患ごとの色になる。
# code 125
wilcox_merge_1 <- list()
for(i in 1:length(wilcox)){
print(paste0("Now processing ", i, " out of ",length(stats_w_normal_2)," proteins"))
wilcox_merge_1[[i]] <- reduce(wilcox[[i]], rbind)
}
names(wilcox_merge_1) <- names(stats_w_normal_2)
# Check size
length(stats_w_normal_2) # 81850
length(wilcox_merge_1) # 81850
stats_w_normal_w_wilcox <- list()
# for(i in 1:5){
# for(i in 1:length(stats_w_normal)){
for(i in 1:length(stats_w_normal_2)){
print(paste0("Now processing ", i, " out of ",length(stats_w_normal_2)," proteins"))
stats_w_normal_w_wilcox[[i]] <- inner_join(stats_w_normal_2[[i]], wilcox_merge_1[[i]])
}
names(stats_w_normal_w_wilcox) <- names(stats_w_normal_2)ここまでの結果をRDataとして外部に保存する。処理にかなりの時間を要するためである。
# code 126
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_6.RData")バイオリンプロットの準備
続いて、バイオリンプロットの表示の準備をする。まず、作成したリストstats_w_normal_w_wilcoxの各要素のデータフレームから、列Tissue_Diseaseに文字列”Tumor_”を含んでいる値を抽出してきて、それらをリストtumor_violinに順次いれていく。これは各タンパク質の発現量もしくは各遺伝子セットのエンリッチメントスコアの中央値をバイオリンプロットと上に水平線として加えるために使用する。
# code 127
tumor_violin <- list()
for(i in 1:length(stats_w_normal_w_wilcox)){
print(paste0("Now processing ", i, " out of ",length(stats_w_normal_2)," genes"))
tumor_violin[[i]] <- stats_w_normal_w_wilcox[[i]] %>% filter(str_detect(Tissue_Disease, "Tumor_") == TRUE)
}
names(tumor_violin) <- names(stats_w_normal_2)関心のあるタンパク質もしくは遺伝子セットの発現量をリストstats_w_normal_w_wilcoxから取得して、データフレームprotein_geneset_dfに入れる。どのタンパク質もしくは遺伝子セットを使用するかは、code 121で作成したデータフレームprotein_geneset_nameを見ながら選んだら良い。
データフレームprotein_geneset_dfを列Disease_Type_Abbreviationと列Tissue_Typeで並べ直し、列Tissue_Diseaseの値の重複を除き、データフレームorder_disease_2を作成する。そして、そのデータフレームorder_disease_2から列Tissue_Diseaseを選んで、その値をベクトルorder_disease_3とする。このベクトルorder_disease_3を使ってデータフレームprotein_geneset_dfの列Tissue_Diseaseにfactor()を設定する。これがバイオリンプロットで表示する際の横軸(疾患)の順番になる。このファクターは、もしかしたらリストstats_w_normal_w_wilcoxの各要素に適応してしまったほうが良いかもしれない。
そして、データフレームprotein_geneset_dfの列名の6列目からタンパク質名もしくは遺伝子セット名を取得して、ベクトルprotein_geneset_show(値が1つのベクトル)を作り、そのベクトルprotein_geneset_showの値を使って、リストtumor_violinから関心のあるタンパク質もしくは遺伝子セットのデータを取得し、データフレームprotein_geneset_df_tumorを作成する。このデータフレームprotein_geneset_df_tumorの6列目、すなわち症例ごとのタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアは、中央値を計算するために使用する。この中央値はバイオリンプロット上に表示する水平線になる。
# code 128
### -------------- run below to show violin plot v.4----------------
### Run following code to show violin plot of the gene or geneset of interest.
#### Specify gene or geneset to be shown.
protein_geneset_df <- stats_w_normal_w_wilcox[["HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva"]] # HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva # CSNK2A1
# ## Add factor
protein_geneset_df <- protein_geneset_df %>% arrange(Disease_Type_Abbreviation, Tissue_Type)
order_disease_2 <- protein_geneset_df %>% distinct(Tissue_Disease)
order_disease_3 <- order_disease_2$Tissue_Disease
protein_geneset_df$Tissue_Disease <- factor(protein_geneset_df$Tissue_Disease,
levels = order_disease_3,
labels = order_disease_3,
exclude = NA, ordered = is.ordered(protein_geneset_df$Tissue_Disease), nmax = NA)
protein_geneset_show <- names(protein_geneset_df)[6]
protein_geneset_df_tumor <- tumor_violin[[protein_geneset_show]]
#### check dataset
view(stats_w_normal_w_wilcox[[protein_geneset_show]])
table(stats_w_normal_w_wilcox[[protein_geneset_show]]$color_2)
table(stats_w_normal_w_wilcox[[protein_geneset_show]]$Tissue_Disease, stats_w_normal_w_wilcox[[protein_geneset_show]]$color_2)バイオリンプロットを表示する
バイオリンプロットの表示にはggplot()を使う。主にデータフレームprotein_geneset_dfを使用する。また、バイオリンプロットのY軸はタンパク質名もしくは遺伝子セット名になり、これは着目するタンパク質もしくいは遺伝子セット毎に変わってしまう。なの4行目にあるようにで、.data[[protein_geneset_show]]のようにデータフレームを動的に参照出来るようにする。これでこのggplot()内ではデータフレームprotein_geneset_df、その中の列もベクトルprotein_geneset_showに入っている値を参照する事が出来る。、そして、列color_2の値に応じてバイオリンプロットの色を変えることにする。色の意味は以下の表の通りである。
| 列 | 値 | 色 | 意味 |
|---|---|---|---|
| color_2 | 0_low | lightblue | 各疾患(がん組織)のタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアの中央値が、全疾患の25パーセンタイル以下である |
| color_2 | 1_even | gray | 各疾患(がん組織)のタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアの中央値が、全疾患の25パーセンタイルより高く、75パーセンタイルより低い。 |
| color_2 | 2_high | red | 各疾患(がん組織)のタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアの中央値が、全疾患の75パーセンタイル以上である。 |
| color_2 | 3_significant | lightgreen | 対象とする臓器のがん組織と正常組織におけるタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアに統計的有意差がある。 |
| color_2 | 4_not_significant | gray | 対象とする臓器のがん組織と正常組織におけるタンパク質発現量もしくは遺伝子セットのエンリッチメントスコアに統計的有意差がない。 |
また、バイオリンプロットには青、黒、赤の水平線を引く。青、黒、赤はそれぞれ各疾患の25パーセンタイル、中央値、75パーセンタイルである。20行目以降は、これをtiffとして保存するためのコードである。tiff()で出力する画像の条件を指定し、以降にggplot()を入れ、最後にdev.off()でtiff()を終了させる。
# code 129
#### show violin plot
ggplot(protein_geneset_df, aes(x = Tissue_Disease, y = .data[[protein_geneset_show]], fill = color_2)) +
geom_violin(width = 1, trim = FALSE) +
geom_hline(yintercept = quantile(protein_geneset_df_tumor[[protein_geneset_show]],
probs = c(0.25, 0.5, 0.75)),
linetype = 1,
color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "red", "lightgreen", "gray")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
#### color of violin plot;
## 0_low; "lightblue"
## 1_even; "gray"
## 2_high; "red"
## 3_significant; "lightgreen"
## 4_not_significant; "gray"
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/", protein_geneset_show, "", ".tiff", sep = "")
tiff(filename = path,
width = 2500, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
ggplot(protein_geneset_df, aes(x = Tissue_Disease, y = .data[[protein_geneset_show]], fill = color_2)) +
geom_violin(width = 1, trim = FALSE) +
geom_hline(yintercept = quantile(protein_geneset_df_tumor[[protein_geneset_show]], probs = c(0.25, 0.5, 0.75)), linetype = 1, color = c("blue", "black", "red"), size = 1) +
scale_fill_manual(values = c("lightblue", "gray", "red", "lightgreen", "gray")) +
theme(axis.text.x = element_text(angle = 90, hjust = 1),
legend.position = "none")
dev.off()
ここまでの結果をRDataで保存する。
# code 130
# save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_6.RData")
各データフレームを他のコードや研究にも利用するために、必要なデータをTSVファイルやRDSファイルで外部に保存する。ついでにここまでの進捗もRDataで外部に保存しておく。
# code 131
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
write_tsv(analysis_5, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/analysis_5.tsv")
write_tsv(analysis_5_tumor, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/analysis_5_tumor.tsv")
saveRDS(stats_w_normal_w_wilcox, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/stats_w_normal_w_wilcox.rds")
saveRDS(tumor_violin, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/tumor_violin.rds")
# This is the dataset for violin plot.
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_6.RData")生存解析
カプランマイヤー法による生存解析を行う。ここではタンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアで症例を階層化し、各疾患ごとの全期間生存率(Overall Survival; OS)求め、ログランク検定で階層間の統計的有意差を求め、関心のあるタンパク質もしくは遺伝子セットが、その患者の生存期間に顕著な影響を与えるかどうかを検討する。
生存解析に用いるデータの準備をする
ここまでの解析でRやRStudioなどの開発環境にかなりのデータが保存されているはずであり、このまま環境にデータを入れていくと計算が遅くなる可能性がある。なので、一旦データを全部クリアする。上記のcode 131で必要なデータは外部に保存しているので、大丈夫なはずである。
データを一旦クリアしたら、今度は生存解析のためのデータを準備する。
code 131で外部に保存したデータフレームanalysis_5_tumorをread_tsv()で読み、それをデータフレームtumorとする。
生存解析に使用する列を確認するために、データフレームtumorの列81852から最終列まで取ってきて、それをデータフレームcolumns_survivalとする。それを見ながら、必要な列を選び、生存解析用にデータを整理する。
まず、データフレームtumorの列Vital_Statusから、値が「Alive」と「Dead」の症例を選んできて、データフレームtumor_2とする。
このデータフレームtumor_2に、全生存期間を入れるための列Days_to_Last_Follow_Up_2を作成する。この時点では全行にNAを入れておく。そして、列Days_to_Last_Follow_Up_2に以下の表の条件で値を入れていく
| データフレーム | 条件1 | 理論演算子 | 条件2 | 入れる値 |
|---|---|---|---|---|
| tumor_2 | 列Vital_Statusが「Alive」 | & | 列Days_to_Deathが「NA」 | 列Days_to_Last_Follow_Upの値 |
| tumor_2 | Vital_Statusが「Dead」 | & | 列Days_to_Last_Follow_Upが「NA」 | 列Days_to_Deathの値 |
| tumor_2 | Vital_Statusが「Dead」 | & | 列Days_to_Deathが「NA」 | 列Days_to_Last_Follow_Upの値 |
| tumor_2 | 上記以外 | – | – | 列Days_to_Last_Follow_Upの値 |
そして、データフレームtumore_2の列Days_to_Last_Follow_Up_2が0よりも長い症例を残し、その列Days_to_Last_Follow_Up_2の列名をDayに変換する。
またこのcode 132の最後に生存解析の打ち切り(イベント発生)の値を入れる列survivalを作成する。データフレームtumor_2の列Vital_Statusの値が「Alive」の場合は列survivalに0(生存)を、値が「Dead」の場合は列survivalに1(死亡により打ち切り)を入れていく。
# code 132
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster.
tumor <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/analysis_5_tumor.tsv")
columns_survival <- colnames(tumor[,81852:ncol(tumor)]) %>% data.frame() %>% rename("column" = ".")
# check case with dead or alive.
table(tumor$Vital_Status)
# Alive Dead Not Reported Unknown
# 782 579 60 2
# case with "Not reported" will be removed.
tumor_2 <- tumor %>% filter(Vital_Status == "Alive" | Vital_Status == "Dead")
# prepare empty column.
tumor_2$Days_to_Last_Follow_Up_2 <- c(rep(NA, nrow(tumor_2))) %>% as.numeric()
for(i in 1:nrow(tumor_2)){
if(tumor_2$Vital_Status[i] == "Alive" & is.na(tumor_2$Days_to_Death[i]) == TRUE){
tumor_2$Days_to_Last_Follow_Up_2[i] <- tumor_2$Days_to_Last_Follow_Up[i]}
else if(tumor_2$Vital_Status[i] == "Dead"& is.na(tumor_2$Days_to_Last_Follow_Up[i]) == TRUE){
tumor_2$Days_to_Last_Follow_Up_2[i] <- tumor_2$Days_to_Death[i]}
else if(tumor_2$Vital_Status[i] == "Dead"& is.na(tumor_2$Days_to_Death[i]) == TRUE){
tumor_2$Days_to_Last_Follow_Up_2[i] <- tumor_2$Days_to_Last_Follow_Up[i]}
else{tumor_2$Days_to_Last_Follow_Up_2[i] <- tumor_2$Days_to_Last_Follow_Up[i]}
}
tumor_2 <- tumor_2 %>% filter(Days_to_Last_Follow_Up_2 > 0)
tumor_2 <- tumor_2 %>% rename("Day" = "Days_to_Last_Follow_Up_2")
tumor_2 <- tumor_2 %>% mutate(
survival = case_when(
Vital_Status == "Alive" ~ 0,
Vital_Status == "Dead" ~ 1
)
)出来上がったデータフレームtumor_2を、今後どこかで利用するために外部に保存する。
# code 133
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
write_tsv(tumor_2, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/tumor_2.tsv")データフレームtumor_2の列Disease_Type_Abbreviationと列case_numberを結合させて列Disease_Type_orderを作成する。そして、以下の表の列を選んできて、データフレームtumor_3を作成する。
| 列 | 列名 | 値 |
|---|---|---|
| 1 | ID_2 | case1323, case1357 … |
| 82120 | Disease_Type_order | LUAD1, LUAD2 … |
| 82121 | Disease_Type_Abbreviation | LUAD, ccRCC … |
| 81901 | Program_Name | Clinical Proteomic Tumor Analysis Consortium … |
| 82125 | Day | 423, 1013 … |
| 82126 | survival | 0, 1 … |
| 2から5209 | タンパク質 | A1BG, A2M … |
| 5210から81851 | 遺伝子セット | chr10p12_escape, chr10p13_escape … |
# code 134
# add new labels for each case.
tumor_2 <- tumor_2 %>% unite(col = Disease_Type_order, Disease_Type_Abbreviation, case_number, sep= "", remove = FALSE)
table(tumor_2$Disease_Type_order)
# just look at the columns.
columns_survival_2 <- colnames(tumor_2[,81852:ncol(tumor)]) %>% data.frame() %>% rename("column" = ".")
# In addition to "ID_2 1", "Disease_Type_order 82120", "Disease_Type_Abbreviation 82121", "Program_Name 81901", Day 82125","survival 82126", choose required columns.
tumor_3 <- tumor_2[,c(1, 82120, 82121, 81901, 82125, 82126, 2:5209, 5210:81851)]作成したデータフレームtumor_3は、今後どこかで利用するために外部に保存する。
# code 135
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
write_tsv(tumor_3, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/tumor_3.tsv")疾患毎の四分位数を計算して階層化する
すでにかなり大量のデータがRに保持されており、計算がかなり遅くなる可能性がある。計算よりも、データをメモリに入れたり、出したり…というのがかなり遅くなるはずであり、結果として、データフレームを開くだけでソフトがクラッシュするし、1つの計算に掛かる時間が増加してしまう。依って、一旦Rに保持しているデータを一旦全部削除(クリア)もしくはRを再起動して、上記で保存したデータフレームtumor_2とデータフレームtumor_3をread_tsv()で読み込んでくる。
# code 136
# # when 2025 10 24 TCGA survival.RData is loaded, memory usage is too big, which is about 800GB, and then the caclulcation is getting very slow. It probably is "swap thrashing" (See this; https://zenn.dev/mabo23/articles/cf7a376b00ed74). Instead of 2025 10 24 TCGA survival.RData (which is containing all dataset in this analysis), load each dataset which will be used for the calculation separately. This mitigate memory usage and speed of calculation is getting faster. Following code is for save each dataset that will be used later.
tumor_2 <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/tumor_2.tsv")
tumor_3 <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/tumor_3.tsv")データフレームtumor_3から、各タンパク質の発現量、もしくは各遺伝子セットのエンリッチメントスコアをリストanalysis_survivalの各要素に入れる。各要素には列Disease_Type_orderなどの分類や列Day、列survivalといった生存解析に必要な値も入れる。
# code 137
# column 1; ID_2; case1323...
# column 82120; Disease_Type_order; LUAD1...
# column 82121; Disease_Type_Abbreviation; LUAD...
# column 81901; Program_Name; Clinical Proteomic Tumor Analysis Consortium...
# column 82124; Day; see the code chunk just above.
# column 82125; survival; see the code chunk just above.
# column 2:5209; gene expression
# column 5210:81851; results of enrichment
analysis_survival <- list()
# view(analysis_3[,c(1:6,7)]) # check them
for(i in 7:ncol(tumor_3)){
print(paste0("Now processing ", i-6, " out of ", ncol(tumor_3)-6, " proteins"))
analysis_survival[[i-6]] <- tumor_3[,c(1:6,i)] %>% data.frame()}
names(analysis_survival) <- colnames(tumor_3[,7:ncol(tumor_3)])リストanalysis_survivalの各要素毎に、タンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアの25パーセンタイル、中央値、75パーセンタイルを算出し、それらをそれぞれ列Q25、列Q50、列Q75に入れる。全行には同じ値が入っているはずである。その値のうち中央値を使って患者を階層化する。階層化に使用した値の範囲は、以下の表の通りである。
| 条件 | 新しく作る列 | 値(階層) |
|---|---|---|
| タンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアの値が、列Q50の値以上 | layer_2 | high |
| タンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアの値が、列Q50の値より低い | layer_2 | low |
| タンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアの値が、列Q75以上の値 | layer_3 | high |
| タンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアの値が、列Q25の値より高く、列Q75の値より低い | layer_3 | mid |
| タンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアの値が、列Q25の値以下 | layer_3 | low |
そして、列layer_2と列layer_3にfactor()を与える。これがグラフ毎に変わっていては見にくくて仕方がないためである。
# code 138
# USE for(), DO NOT USE foreach(). foreach() spent huge amount of memory, which was more than 1 TB, and it took very long time to store the data for the execution.
for(i in 1:length(analysis_survival)){
print(paste0("Now processing ", i, " out of ", length(analysis_survival), " genes"))
# IMPORTANT; Need to use ".data[[]]" not "temp_survival_df"
gene_name_survival <- names(analysis_survival[[i]][7])
temp_survival_df <- analysis_survival[[i]] %>% data.frame()
temp_survival_quantile <- temp_survival_df %>% group_by(Disease_Type_Abbreviation) %>%
summarise(Q50 = quantile(.data[[gene_name_survival]], probs = 0.50, na.rm = TRUE),
Q25 = quantile(.data[[gene_name_survival]], probs = 0.25, na.rm = TRUE),
Q75 = quantile(.data[[gene_name_survival]], probs = 0.75, na.rm = TRUE),
n = n())
## color matching
# color_matching <- match(analysis$disease, stats_EMT$disease)
# analysis$color <- stats_EMT$color[color_matching]
disease_matching <- match(temp_survival_df$Disease_Type_Abbreviation, temp_survival_quantile$Disease_Type_Abbreviation)
temp_survival_df$Q50 <- temp_survival_quantile$Q50[disease_matching]
temp_survival_df$Q25 <- temp_survival_quantile$Q25[disease_matching]
temp_survival_df$Q75 <- temp_survival_quantile$Q75[disease_matching]
analysis_survival[[i]] <- analysis_survival[[i]] %>%
mutate(
Q25 = temp_survival_df$Q25,
Q50 = temp_survival_df$Q50,
Q75 = temp_survival_df$Q75,
layer_2 = case_when(analysis_survival[[i]][[gene_name_survival]] >= temp_survival_df$Q50 ~ "high",
analysis_survival[[i]][[gene_name_survival]] < temp_survival_df$Q50 ~ "low"),
layer_3 = case_when(analysis_survival[[i]][[gene_name_survival]] >= temp_survival_df$Q75 ~ "high",
analysis_survival[[i]][[gene_name_survival]] > temp_survival_df$Q25 & analysis_survival[[i]][[gene_name_survival]] < temp_survival_df$Q75 ~ "mid",
analysis_survival[[i]][[gene_name_survival]] <= temp_survival_df$Q25 ~ "low")
)
# set order for layer_2
analysis_survival[[i]]$layer_2 <-
factor(analysis_survival[[i]]$layer_2,
levels = c("high", "low"),
labels = c("high", "low"),
exclude = NA, ordered = is.ordered(analysis_survival[[i]]$layer_2), nmax = NA)
# set order for layer_3
analysis_survival[[i]]$layer_3 <-
factor(analysis_survival[[i]]$layer_3,
levels = c("high", "mid", "low"),
labels = c("high", "mid", "low"),
exclude = NA, ordered = is.ordered(analysis_survival[[i]]$layer_3), nmax = NA)
}
# check data
view(analysis_survival[[2]])
table(analysis_survival[[1]]$layer_2)
table(analysis_survival[[1]]$layer_3)カプランマイヤー法により生存期間を計算する
リストanalysis_survivalの各要素名を取得し、データフレームprotein_name_survival_2を作成する。これは、生存解析に使用するタンパク質もしくは遺伝子セットを選ぶためのデータフレームである。それを眺めて、生存解析に使用するタンパク質もしくは遺伝子セットを選んでくる。ここでは、「HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva」を選ぶ。選んだら、そのタンパク質もしくは遺伝子セットをベクトルprotein_geneset_interestに入れる。
次に、リストanalysis_survivalの何らかの要素から(どれでもいい。どれでも同じ疾患が入っているはず)疾患名を取得し、それをベクトルdisease_nameに入れる。
# code 139
protein_name_survival_2 <- data.frame(protein_geneset = names(analysis_survival)) # for looking up gene or geneset of interest
view(protein_name_survival_2) # look for gene or geneset of interest
protein_geneset_interest <- "HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva" # put gene or geneset of interest for survival curve
# HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva
# CSNK2A1
#
disease_name <- analysis_survival[[1]]$Disease_Type_Abbreviation %>% unique() %>% data.frame() # any number is OK.
colnames(disease_name) = "disease"
生存曲線を表示する
リストanalysis_survivalから、上記で選んだタンパク質もしくは遺伝子セットのデータフレームを、ベクトルprotein_geneset_interest(長さ1のベクトル)に入っている値(ここでは「HALLMARK_INTERFERON_GAMMA_RESPONSE_gsva」が入っている)を選び、そのデータフレームを列Disease_Type_Abbreviationの値を使って、リストinterest_protein_geneset_survival_1の各要素に疾患毎に入れていく。そのリストinterest_protein_geneset_survival_1には疾患名をつけておく。
リストinterest_protein_geneset_survival_1の各要素で、生存期間解析を行う。Surv()は生存期間に死亡もしくは打ち切りなのか生存なのかをベクトルとして取得する関数で、この結果得られるオブジェクトをsurvival objectというらしい。survfit()はSurv()で取得した打ち切り入の生存期間(survival object)を使ってカプランマイヤー法により生存曲線を計算する関数である。この結果を疾患毎に行い、それぞれの結果をリストfit_status_1の各要素に入れていく。リストfit_status_1の各要素と、それに対応するリストinterest_protein_geneset_survival_1の各要素をggsurvplot()にいれて、生存曲線を出力する。これが列layer_2の値ごとの生存曲線である。ggsurvplot()はログランク検定の結果も出力する。ちなみにログランク検定は、群間に差があるかどうかを検定する方法であり、どの群で差があるのかはわからないはずである。なので、基本は2群でログランク検定するほうが良いと思う。もし多群ならば、FDR(False Discovery Ratio)の補正などを行う必要がある。
# code 140
# prepare list
interest_protein_geneset_survival_1 <- list()
for(i in 1:11){
interest_protein_geneset_survival_1[[i]] <- analysis_survival[[protein_geneset_interest[1]]] %>% filter(Disease_Type_Abbreviation == disease_name$disease[i])
} # If length(disease_name) was used, the all 33 project were put in the list. 18 means only 18 projects were put in the list.
names(interest_protein_geneset_survival_1) <- disease_name$disease[1:11]
fit_status_1 <- list() # 1 - 11
survival_curve_status_1 <- list() # 1 - 11
length(interest_protein_geneset_survival_1) # 11 diseases
for(i in 1:length(interest_protein_geneset_survival_1)){
fit_status_1[[i]] <- survfit(Surv(Day, survival) ~ layer_2, data = interest_protein_geneset_survival_1[[i]])
survival_curve_status_1[[i]] <- ggsurvplot(
fit_status_1[[i]],
data = interest_protein_geneset_survival_1[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6),
font.legend = c(6),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i])
} # legend = c("right", "left", "top", "bottom", "none") can show which color is which layer.
# red; high
# green; low
survival_curve_status_1_graph <- lapply(survival_curve_status_1, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_1_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/", protein_geneset_interest,"_2_layer_1",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_1_graph, ncol = 6)
dev.off()
code 140は列layer_2(階層が2つ)を使った生存曲線である。以下のcode 141はlayer_3(階層が3つ)を使った生存曲線である。上述の通り、ログランク検定ではどの群に有意差があるのかわからないため、3群ならばそのペアで検定して、そのp値をFDR補正する必要がある。
# code 141
fit_status_3 <- list() # 1 - 15
survival_curve_status_3 <- list() # 1 - 15
length(interest_protein_geneset_survival_1) # 18 diseases
for(i in 1:length(interest_protein_geneset_survival_1)){
fit_status_3[[i]] <- survfit(Surv(Day, survival) ~ layer_3, data = interest_protein_geneset_survival_1[[i]])
survival_curve_status_3[[i]] <- ggsurvplot(
fit_status_3[[i]],
data = interest_protein_geneset_survival_1[[i]],
pval = TRUE,
pval.method = FALSE,
pval.size = 3.5,
risk.table = TRUE,
font.main = c(6, "bold", "black"),
font.x = c(6, "plain", "black"),
font.y = c(6, "plain", "black"),
font.tickslab = c(6, "plain", "black"),
font.legend = c(6, "plain", "black"),
surv.plot.height = 1.0,
legend = "none") + labs(title = disease_name$disease[i])
}# legend = "bottom") can show which color is which layer.
# red; high
# green; mid
# blue; low
survival_curve_status_3_graph <- lapply(survival_curve_status_3, function(x) x$plot)
grid.arrange(grobs = survival_curve_status_3_graph, ncol = 6)
# output
path <- paste0("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/", protein_geneset_interest,"_3_layer_1",".tiff", sep = "")
tiff(filename = path,
width = 1800, height = 800, units = "px", pointsize = 12,
compression = "none",
bg = "white", res = 150, family = "",
symbolfamily="default")
grid.arrange(grobs = survival_curve_status_3_graph, ncol = 6)
dev.off()
ログランク検定により階層間の生存期間の差を検定する
この解析のように、複数の疾患や症例にわたる網羅的なスクリーニングのような解析ならば、まずは階層間に有意差がある疾患だけを拾ってきて、そこから詳しく解析していく方が良い可能性はある。そういう場合は、全疾患に渡るログランク検定の結果をデータフレームにした方がわかりやすいはずである。
まずはログランク検定の結果を入れるリストtemp_logrank_1とリストlogrankを用意する。リストtemp_logrank_1の各要素には、疾患ごとのタンパク質の発現量もしくは遺伝子セットのエンリッチメントスコアを入れていき、それを、更にタンパク質もしくは遺伝子セット毎にリストlogrankの各要素にいれる。要するに、生存解析に必要な値をタンパク質もしくは遺伝子セット毎にわけ、更に疾患毎に分けた。
# code 142
# prepare dataset
temp_logrank_1 <- list()
logrank <- list()
for(x in 1:length(analysis_survival)){
print(paste0("Now processing ", x, " out of ", length(analysis_survival), " genes"))
for(y in 1:length(disease_name$disease)){
temp_logrank_1[[y]] <- analysis_survival[[x]] %>% data.frame() %>% filter(Disease_Type_Abbreviation == disease_name$disease[y])
}
names(temp_logrank_1) <- disease_name$disease
logrank[[x]] <- temp_logrank_1
}
names(logrank) <- names(analysis_survival)
そして、ログランク検定の結果を入れるリストlogrank_pvalと、途中の計算を入れるデータフレームtemp_dataframeを用意する。データフレームtemp_dataframeのそれぞれ列layer_2_pvalと列layer_3_pvalに、各疾患毎にカプランマイヤー法で生存解析し、列layer_2と列layer_3で階層化したときのログランク検定結果を入れていく。
ログランク検定は、生存曲線を示したときと同様にsurvfit(Surv())を用いる。しかしながら、階層化できなくてログランク検定出来ない場合もある。そのときはエラーが出るが、もしエラーがでたらそのときの結果をNAとするようにtryCatch()する。データフレームtemp_dataframeには疾患毎のp値が入っているので、それを今度はリストlogrank_pvalの各要素に、つまり、タンパク質もしくは遺伝子セット毎の結果として入れていく。
# code 143
# calculate log-rank for all disease over all gene and geneset
temp_dataframe <- data.frame(disease = disease_name$disease, layer_2_pval=rep(NA, 11), layer_3_pval=rep(NA, 11))
logrank_pval <- list()
# if analysis_survival is removed because of too big, use length_analysis_survival and names_analysis_survival
length_analysis_survival <- length(analysis_survival)
names_analysis_survival <- names(analysis_survival)
for(x in 1:length(analysis_survival)){
print(paste0("Now processing ", x, " out of ", length(analysis_survival), " genes"))
for(y in 1:length(disease_name$disease)){
temp_df <- logrank[[x]][[y]] %>% data.frame()
temp_dataframe$layer_2_pval[y] <- tryCatch({survdiff(Surv(Day, survival) ~ layer_2, data = temp_df)$p}, error = function(e) {NA})
temp_dataframe$layer_3_pval[y] <- tryCatch({survdiff(Surv(Day, survival) ~ layer_3, data = temp_df)$p}, error = function(e) {NA})
}
logrank_pval[[x]] <- temp_dataframe
}
names(logrank_pval) <- names(analysis_survival)また、多くのタンパク質もしくは遺伝子セットの結果があるので、年のためにFDRの補正を行った後のp値を計算してき、それを列layer_2_pval_FDR及び列layer_3_pval_FDRに入れる。ここでもtryCatch()を使用して、エラーが出たらNAを入れるようにする。そして、リストlogrank_pvalの各要素をreduce()とbind_rows()で、縦方向(行方法)に結合し、データフレームlogrank_pval_dfを作成する。
# code 144
# FDR correction and name the each column
for(i in 1:length(logrank_pval)){
print(paste0("Now processing ", i, " out of ", length(logrank_pval), " genes"))
logrank_pval[[i]] <- logrank_pval[[i]] %>% mutate(
layer_2_pval_FDR = tryCatch({p.adjust(layer_2_pval, method = "fdr")}, error = function(e) {NA}),
layer_3_pval_FDR = tryCatch({p.adjust(layer_3_pval, method = "fdr")}, error = function(e) {NA}),
gene_geneset = names(logrank_pval[i])
)
}
# get the list together to a data frame
logrank_pval_df <- reduce(logrank_pval, bind_rows) %>% data.frame()データフレームlogrank_pval_dfには、どのタンパク質もしくは遺伝子セットが統計的優位に生存に関与するかが、疾患毎に書かれている。エクセル等でざっと見ても良いのだが、行数がかなり多いので、エクセルでは全部を見ることが出来ない。それでRであれば、みたい症例について柔軟に見ることが出来るはずである。ここではその一例として、膵臓がん(PAAD)について見ることにする。
まず、疾患名を取得してきてデータフレームdisease_interestに入れる。このデータフレームdisease_interestの値を使って、患者の生存率に統計的有意な影響を与えるだろうタンパク質もしくは遺伝セットを取得してきて、リストdisease_significant_layer_2の各要素に疾患毎にいれていく。リストdisease_significant_layer_2は列layer_2の階層間での検定結果を、リストdisease_significant_layer_3は列layer_3の階層間での検定結果を入れていく。そして、その2つをそれぞれ、リストdisease_significantの各要素にそれぞれ入れていく。
# code 145
# example; which genes and geneset are closely associated the survival of PAAD patients??
# PAAD_significant <- logrank_pval_df %>% filter(disease == "PAAD" & (layer_2_pval_FDR < 0.05))
disease_interest <- data.frame(table(logrank_pval_df$disease))$Var1
disease_significant_layer_2 <- list()
for(i in 1:length(disease_interest)){
disease_significant_layer_2[[i]] <- logrank_pval_df %>% filter(disease == disease_interest[i] & (layer_2_pval_FDR < 0.05))
}
names(disease_significant_layer_2) <- disease_interest
disease_significant_layer_3 <- list()
for(i in 1:length(disease_interest)){
disease_significant_layer_3[[i]] <- logrank_pval_df %>% filter(disease == disease_interest[i] & (layer_3_pval_FDR < 0.05))}
names(disease_significant_layer_3) <- disease_interest
disease_significant <- list()
disease_significant[["layer_2"]] <- disease_significant_layer_2
disease_significant[["layer_3"]] <- disease_significant_layer_3
これで生存解析は一通り終了である。今後使用するかもしれないオブジェクトをそれぞれRDSファイルやtsvファイルとして外部に保存する。
# code 146
saveRDS(logrank, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/logrank.rds")
saveRDS(logrank_pval, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/logrank_pval.rds")
write_tsv(logrank_pval_df, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/logrank_pval_df.tsv")
saveRDS(disease_significant, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/disease_significant.rds")また、これまでの結果をRDataとして外部に保存する。なお2026 01 10 CPTAC expression matrix_7.RDataはこの解析全体、2026 01 10 CPTAC expression matrix_7_survival_and_logrank.RDataは生存解析からログランク検定までのデータである。Rののコードをもう一度流すよりは、このように全体を保存しておくほうが早く作業を始めることができる。
# code 147
# code 147
# This is all dataset of this analysis.
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_7.RData")
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_7_survival_and_logrank.RData") # only for survival analysis and logrank test. 最後に使用したパッケージなどの情報を日付付きで外部に保存する。
# code 148
file.remove("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/save_log.txt")
file.remove("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/sessionInfo.txt")
cat(paste0("The analysis was saved at ", Sys.time(), "\n"),
file = "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/save_log.txt")
sink("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/sessionInfo.txt")
sessionInfo()
sink()タンパク質発現量と遺伝子発現量の相関を解析する
ここで、話はすこしそれて、同一症例におけるタンパク質発現量と遺伝子発現量の相関を解析する。
CPTACの一部の症例はTCGAにも使用されていることから同一症例におけるタンパク質と遺伝子発現の相関の解析にも使用出来る。このタンパク質と遺伝子発現の相関はあるのか?という問は、素人みたいな奴等からよく聞かれる質問の1つであると、個人的に思っている。個人的にはこの質問はかなりのナンセンスと思う。まず、この質問をしてくる奴等には、なぜタンパク質発現プロファイルを使っているのか、なぜ遺伝子発現プロファイルを使っているのか、その解析の目的は何か等、その解析の性質のようなものを理解していないように思う。大学の研究機関でこれをやったら、結局有用なデータを得ることが出来ずにデータが無駄になるし、企業であれば、結果としてその解析が有用なのかを決定する能力がない証拠である。
個人的には「遺伝子発現とタンパク質発現はある程度相関するものであり、一部のタンパク質の発現はその遺伝子発現との相関は弱いので注意が必要という観点でトランスレーションを進めるべき」と思う。なぜならば、遺伝子発現量が多ければタンパク質発現量もそれなりに多い、という一般的な前提があるためだ。それを無視してこのように「遺伝子発現量とタンパク質発現量に相関はあるのか」ということから始めると、当然そうではないタンパク質も沢山あるので、ある分子が生存期間に与える影響から新しい分子標的を見つける、という目的は、このような網羅的な発現プロファイルを用いている場合はいつになっても達成できないことになる。この疑問を持つなら、最初からCPTACのタンパク質発現プロファイルやTCGAの遺伝子発現プロファイルのどちらか、自分たちの研究や開発において重要な方(タンパク質なのか遺伝子なのか)を理解してから解析をデザインすべきである。また、当然ながらタンパク質発現と遺伝子発現の相関なんて、臓器、組織、疾患、がんのステージなどの様々な状態毎に異なってくるはずである。だから、やはり「遺伝子発現量とタンパク質発現量に相関はあるのか」という質問が単発で出てくる場合、そいつは勉強不足のやつなのかもしれない。基礎研究や創薬の開発の時点で、対象とする疾患では、ステージの後期でタンパク質発現量と遺伝子発現量が相関し、これらはその疾患の生存期間に関わるから….という流れが正しいのかもしれない。逆に、「遺伝子発現量とタンパク質発現量に相関はあるのか」という質問は、結果から解析の前提の検討に戻るような質問とも言える。その一方で、聞かれることも事実である。ということは、一度解析をしておけば流用も出来るだろう、ということである。なので、せっかくダウンロードしてデータの整えたので、やることはやってしまう。
そもそも遺伝子発現量とタンパク質発現量の解析は必要なのか
この相関を出さなければならないという野郎は、専門家や医者だったとしても生物学の研究をまともに行ったことがない輩か。規制当局やレギュラトリーサイエンスをかじっている野郎ではないかと思う。個人的には、このような野郎に多少の発現力があって、そいつが必要と言い張っているとしても、タンパク質と遺伝子発現の相関の解析は一般的には不要だと思う。遺伝子発現量とタンパク質発現量がある程度相関することは、一般的な実験でも明確な根拠があるためである。例えば、siRNAで遺伝子発現をノックダウンさせた場合は、やはりそのタンパク質の発現は大幅に下がる。つまり、siRNAの実験は、遺伝子発現量とタンパク質発現量は相関することが一般的に成り立つことが前提で、実施されるわけである。それに、この解析の最後に論文を引用するが、その論文では遺伝子発現量とタンパク質発現量はある程度相関することが示されている。なので、今更こういったことをグダグダと言っている野郎は、もしかしたらアタマに柔軟がなく、すなわちまともに研究した経験(少なくともまともな論文に研究成果を掲載した経験)がなく、依って、規制当局や投資家などから投げつけられる質問に柔軟に対応出来ない野郎である可能性がある。プロだったら、こういった生物現象の前提をなんかを敢えてデータとして示さずに、何とかするべきである。逆にそれが出来ない野郎はプロではなく、ただ仕事に慣れただけの野郎である。だから、自分は、何かのバリデーションのために間接的な結果(それが直接必要ない結果という意味)として遺伝子発現量とタンパク質発現量の相関を示す必要は無いと考えている。もちろん、その相関が、対象とするメカニズムに直接関わる場合は必要だろう。
データを準備する(Rのコード1に戻る)
ここで、エンリッチメント解析を行う前の、タンパク質発現プロファイル名や各症例の臨床情報の整理を行ったときのファイル(ここから使用するRのダウンロード1)のcode 62に戻って解析を始める。
まずデータフレームfile_name_case_idとデータフレームbiospeciemenを使って、タンパク質と遺伝子の発現量の相関の解析に使えそうなデータを選んでいく。データフレームfile_name_case_idは、code 41で作成したデータフレームであり、missForestで欠損値を補完したタンパク質発現プロファイルresult_missForest_pre_argu_df_median_normalized_dfの列名である。データフレームbiospeciemenは、タンパク質発現プロファイルと各症例の情報を記録したものであり、code 38で作成されている。まず、データフレームbiospeciemenの列Aliquot_Submitter_ID_2の値のうち、重複している値を取ってきてデータフレームcheck_biospeciemen_Aliquot_Submitter_ID_2とする。同様に、データフレームfile_name_case_idからは列possible_Aliquot_Submitter_IDの値のうち、重複している値を取ってきてデータフレームcheck_file_name_case_id_possible_Aliquot_Submitter_IDを作成する。まず、データフレームbiospeciemenの列Aliquot_Submitter_ID_2は、列Aliquot_Submitter_IDの値のハイフンをアンダーバーに置き換えたものである。これはタンパク質発現プロファイル名と一致するはずである。また、データフレームfile_name_case_idの列possible_Aliquot_Submitter_IDは、タンパク質発現プロファイル名から列Aliquot_Submitter_IDの値として使用されている部分を抽出してきた値が保持されている。これらを一致させることで、データフレームbiospeciemenとデータフレームfile_name_case_idを結合させることが出来る。しかしながら、これらの列には、なんと重複している値が存在している。この意味は、「1つのタンパク質発現プロファイルが、複数の症例で得られている」ということであり、それはおかしい。また、この解析では、「1つのタンパク質発現プロファイルは、1つの症例に関連付けられていれば良い」はずである。なので、上記の通り、データフレームbiospeciemenの列Aliquot_Submitter_ID_2、データフレームfile_name_case_idの列possible_Aliquot_Submitter_IDの重複は除いてしまう。これによりデータフレームbiospeciemen_corとデータフレームfile_name_case_id_corを作成する。また、後の処理をなるべく簡単にするように、それぞれの列名をAliquot_Submitter_ID_corに変更し、おかしな重複がないことを確認する。
確認のために、データフレームfile_name_case_id_corとデータフレームbiospeciemen_corを、列Aliquot_Submitter_ID_corでinner_join()して、データフレームclinical_for_correlationを作成する。このデータフレームclinical_for_correlationの症例数は、2994例らしい。ただし、inner_join()で結合するのは、考えるとこれは少し危ないように思う。もし、同一症例で別のタンパク質発現プロファイルが採られていたらどうだろうか….どちらかが間違って関連付けられる可能性がある。ということで、このデータフレームclinical_for_correlationは確認用だけで、解析には使用しない。
データフレームfile_name_case_id_corとデータフレームbiospeciemen_corを関連付けるためには、ここはアナログ的力技を用いる。71行目から85行目では、空のベクトルcase_IDを用意し、そこに、データフレームfile_name_case_id_corの列Aliquot_Submitter_ID_corと、データフレームbiospeciemen_corの列Aliquot_Submitter_ID_corが一致している行の、データフレームbiospeciemen_corの列Case_IDの値を入れていく。これで、データフレームfile_name_case_id_corの列Aliquot_Submitter_ID_corに対応するデータフレームbiospeciemen_corの列Case_IDの値を得ることが出来るはずである。ちなみに、このfor(){}のなかでbreakを使っているが、これを使わなくても結果は同じだった。そして、得られたベクトルCase_IDをデータフレームfile_name_case_id_corに列Case_IDとして加えておいた。これらは後から使用するかもしれないので、TSVファイルとして外部に保存しておいた。
# code 62
# Running this code chunk will take 4 - 5 min.
# "expression" stores column name of mass spec count data (which is file name of mass spec count data).
# "biospeciemen" stores data of "PDC_biospecimen_manifest_07292024_100158.tsv". It is same as column name (which is file name of mass spec count data).
# It means that file name of mass spec data links to Case ID for matching Primary Site (like Ovary, Breast...), Disease Type (like Lung Adenocarcinoma, Glioblastoma...) at latter analysis.
# Following code retrieve "Case ID" of the mass spec count data.
# Strings in biospeciemen$"Aliquot Submitter ID" contains expression$possible_file_name
nrow(file_name_case_id) # 5202
nrow(biospeciemen) # 5228
# check duplication
## If these values were duplicated, it will be a problem. If the value is duplicated, the value was probably linked to different cases. These values are the name of protein expression profile and therefore they need to be unique.
check_biospeciemen_Aliquot_Submitter_ID_2 <- table(biospeciemen$Aliquot_Submitter_ID_2) %>% data.frame() %>% rename("Aliquot_Submitter_ID_2" = "Var1") %>% filter(Freq >1)
check_file_name_case_id_possible_Aliquot_Submitter_ID <- table(file_name_case_id$possible_Aliquot_Submitter_ID) %>% data.frame() %>% rename("possible_Aliquot_Submitter_ID" = "Var1") %>% filter(Freq >1)
# Remove duplication
biospeciemen_cor <- biospeciemen[biospeciemen$Aliquot_Submitter_ID_2 %in% check_biospeciemen_Aliquot_Submitter_ID_2$Aliquot_Submitter_ID_2 == FALSE,]
file_name_case_id_cor <- file_name_case_id[file_name_case_id$possible_Aliquot_Submitter_ID %in% check_file_name_case_id_possible_Aliquot_Submitter_ID$possible_Aliquot_Submitter_ID == FALSE,]
# Change column name of biospeciemen_cor$AAliquot_Submitter_ID_2 and file_name_case_id_cor$possible_Aliquot_Submitter_ID into "Aliquot_Submitter_ID_cor"
biospeciemen_cor <- biospeciemen_cor %>% rename("Aliquot_Submitter_ID_cor" = "Aliquot_Submitter_ID_2")
file_name_case_id_cor <- file_name_case_id_cor %>% rename("Aliquot_Submitter_ID_cor" = "possible_Aliquot_Submitter_ID")
# Before merging, check duplication.
check_biospeciemen_cor_Aliquot_Submitter_ID_cor <- table(biospeciemen_cor$Aliquot_Submitter_ID_cor) %>% data.frame() %>% rename("Aliquot_Submitter_ID_cor" = "Var1") %>% filter(Freq >1) # There is no duplication
check_file_name_case_id_cor_Aliquot_Submitter_ID_cor <- table(file_name_case_id_cor$Aliquot_Submitter_ID_cor) %>% data.frame() %>% rename("Aliquot_Submitter_ID_cor" = "Var1") %>% filter(Freq >1) # There is no duplication
nrow(biospeciemen_cor) # 5127 case
nrow(file_name_case_id_cor) #4916 case
table(biospeciemen_cor$Aliquot_Submitter_ID_cor %in% file_name_case_id_cor$Aliquot_Submitter_ID_cor)
# FALSE TRUE
# 2133 2994
# (total is 5127)
# The data frame "clinical_for_correlation" did not used in this analysis.
clinical_for_correlation <- inner_join(file_name_case_id_cor, biospeciemen_cor, by = "Aliquot_Submitter_ID_cor", suffix = c("", "_biospeciemen"))
nrow(clinical_for_correlation) # 2994 cases
## I feel something weird in following code. I will use "clinical_for_correlation", which is prepared with inner_join().
# # Create an empty vector to store the results
# Case_ID <- character(nrow(file_name_case_id))
# for (x in 1:nrow(file_name_case_id_cor)) {
# for (y in 1:nrow(biospeciemen_cor)) {
# if (grepl(file_name_case_id_cor$possible_Aliquot_Submitter_ID[x], biospeciemen_cor$Aliquot_Submitter_ID_2[y])) {
# Case_ID[x] <- biospeciemen_cor$Case_ID[y]
# break
# }
# }
# }
# Create an empty vector to store the results
Case_ID <- character(nrow(file_name_case_id_cor))
# biospeciemen_cor holds 5127 cases
# file_name_case_id_cor hoids 4916 cases
# shared value is 2994 cases
# each data frame does not have duplication in column "Aliquot_Submitter_ID_cor".
for (x in 1:nrow(file_name_case_id_cor)) {
for (y in 1:nrow(biospeciemen_cor)) {
if (grepl(file_name_case_id_cor$Aliquot_Submitter_ID_cor[x], biospeciemen_cor$Aliquot_Submitter_ID_cor[y])) {
Case_ID[x] <- biospeciemen_cor$Case_ID[y]
break
}
}
}
length(Case_ID) # 4916 case
#Then, Add the Case ID into the colume name (which is fle name of each mass spec count data).
file_name_case_id_cor$Case_ID <- Case_ID
view(file_name_case_id_cor)
write_tsv(file_name_case_id_cor, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/file_name_case_id_matching.tsv")ここでは、相関の解析に使用するタンパク質発現プロファイルを選んでくる。
データフレームfile_name_case_id_corの列Case_IDは、上記のcode 62で値が一致しなかった行に関しては空欄が入力されている。なので、そのデータフレームfile_name_case_id_corの列Case_IDが空欄の例は除いておく。そしてそれをデータフレームsample_with_case_idとする。
そして、タンパク質発現プロファイルであるデータフレームresult_missForest_pre_argu_df_median_normalized_dfから、データフレームfile_name_case_id_corの列name(タンパク質発現プロファイル名)で、一致するタンパク質発現プロファイルを抽出して、データフレームresult_missForest_pre_argu_df_median_normalized_df_matched_case_IDを作成する。
その行(タンパク質)と列(タンパク質発現プロファイル)数を確認すると、それぞれ2994例、5208タンパク質だった….データフレームclinical_for_correlationと同じだ….なんてこった。まあいいか。確認できたということで。25行目と26行目で列Case_IDの重複を確認しているが、この列の重複は、許容する。1つの症例から、複数のタンパク質発現プロファイルを得ているということで、これは有り得ることのはず。
データフレームresult_missForest_pre_argu_df_median_normalized_df_matched_case_IDは、後ほど使用するので、行名を列proteinに直してデータフレームresult_missForest_pre_argu_df_median_normalized_df_matched_case_ID_outputを作成し、これをTSVファイルとして保存しておく。
# code 63
# Remove empty value at Case_ID column in "sample", which is the file names of mass spec count data.
# sample_with_case_id <- clinical_for_correlation
# sample_with_case_id$name
sample_with_case_id <- file_name_case_id_cor %>% filter(Case_ID != "")
sample_with_case_id$name
# sample_with_case_id <- sample_with_case_id %>% rename("Case ID" = "Case_ID")
result_missForest_pre_argu_df_median_normalized_df_matched_case_ID <-
result_missForest_pre_argu_df_median_normalized_df %>% dplyr::select(all_of(sample_with_case_id$name))
# check size of data frame.
ncol(result_missForest_pre_argu_df_median_normalized_df_matched_case_ID) # 2994
nrow(result_missForest_pre_argu_df_median_normalized_df_matched_case_ID) # 5208
dim(result_missForest_pre_argu_df_median_normalized_df_matched_case_ID) # [1] 5208 2994
# 5208 genes and 2994 patients were remained.
check_Case_ID <- table(sample_with_case_id$Case_ID) %>% data.frame() %>% filter(Freq > 1)
check_Case_ID_2 <- sample_with_case_id %>% group_by(Case_ID) %>% filter(n() > 1)
result_missForest_pre_argu_df_median_normalized_df_matched_case_ID_output <- result_missForest_pre_argu_df_median_normalized_df_matched_case_ID %>% rownames_to_column(var = "protein")
write_tsv(result_missForest_pre_argu_df_median_normalized_df_matched_case_ID_output, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/result_missForest_pre_argu_df_median_normalized_df_matched_case_ID_output.tsv")ここでは、タンパク質発現プロファイル名とそれに対応するCase IDを持つデータフレームfile_name_case_id_corと、biospeciemenの情報を結合させる。
これまでの処理で出来たデータフレームと区別するために、データフレームfile_name_case_id_corをデータフレームsample_of_PDC_for_TCGAとしてコピーする。また、改めてbiospecimemenの情報を読み込んできて、データフレームPDC_biospecimen_manifestとする。データフレームPDC_biospecimen_manifestの列名の空欄をアンダーバーに変更しておく。
次に、データフレームPDC_biospecimen_manifestの列Case_IDでの値の重複を除去して、データフレームPDC_biospecimen_manifest_distinctを作成する。これはタンパク質発現プロファイルの取得に使用された試料の情報であり、また、すでにデータフレームsample_of_PDC_for_TCGAに列Case_IDは付いている。なので、このデータフレームデータフレームPDC_biospecimen_manifestで必要なのは、一致させるかどうかではなく、単にその列Case_IDの他の臨床情報である。列Case_IDの重複を除去して、そのCase_IDが残っていれば、その情報をデータフレームsample_of_PDC_for_TCGAに結合することが出来る。そして、データフレームPDC_biospecimen_manifestに、列Aliquot_Submitter_IDの値のハイフンをアンダーバーに変えた列Aliquot_Submitter_ID_corを新しく作成しておく。この列Aliquot_Submitter_ID_corと列Case_IDを使って、データフレームsample_of_PDC_for_TCGAとデータフレームPDC_biospecimen_manifest_distinctをinner_join()し、データフレームsample_of_PDC_for_TCGA_with_biospecimen_manifestを作成する。これで、使用できそうなタンパク質発現プロファイル名に、一致する試料の臨床情報を結合することが出来る。
# code 64
# "sample_of_PDC_for_TCGA" contains file name of mass spec count data and their Case ID to match clinical info.
# sample_of_PDC_for_TCGA <- file_name_case_id %>% rename("Case ID" = "Case_ID")
sample_of_PDC_for_TCGA <- file_name_case_id_cor
# Need to connect E drive.
PDC_biospecimen_manifest <- read_tsv(file= "/mnt/seqdata/public_data/Blog/CPTAC/PDC_biospecimen_manifest_10202024_102342.tsv")
# Replace " " into "_" in columns of "PDC_biospecimen_manifest".
colnames_PDC_biospecimen_manifest <- gsub(pattern = " ", replacement = "_", x = colnames(PDC_biospecimen_manifest))
colnames(PDC_biospecimen_manifest) <- colnames_PDC_biospecimen_manifest
PDC_biospecimen_manifest_distinct <- PDC_biospecimen_manifest %>% dplyr::distinct(Case_ID, .keep_all = TRUE)
PDC_biospecimen_manifest_distinct <- PDC_biospecimen_manifest_distinct %>% mutate(
Aliquot_Submitter_ID_cor = gsub(pattern = "-", replacement = "_", x = Aliquot_Submitter_ID)
)
PDC_biospecimen_manifest_distinct <- PDC_biospecimen_manifest_distinct %>% select(
c(1,2, ncol(PDC_biospecimen_manifest_distinct), 3:(ncol(PDC_biospecimen_manifest_distinct)-1))
)
# Merge "sample_of_PDC_for_TCGA" with "PDC_biospecimen_manifest_distinct" to get clinical information for each sample.
# sample_of_PDC_for_TCGA_with_biospecimen_manifest <- full_join(
# sample_of_PDC_for_TCGA,
# PDC_biospecimen_manifest_distinct,
# by = Case_ID,
# suffix = c("_sample_of_PDC_for_TCGA", "_PDC_biospecimen_manifest_distinct"))
nrow(sample_of_PDC_for_TCGA) # 4916 cases; sample_of_PDC_for_TCGA holds name of protein expression profile.
nrow(PDC_biospecimen_manifest_distinct) # 3523 cases; sample_of_PDC_for_TCGA holds information of speciemens or samples that were used for MS.
# PDC_biospecimen_manifest_distinct$Aliquot_Submitter_ID_cor
# sample_of_PDC_for_TCGA$Aliquot_Submitter_ID_cor
colnames(sample_of_PDC_for_TCGA) # Needs "Aliquot_Submitter_ID_cor"
colnames(PDC_biospecimen_manifest_distinct) # "Needs "Aliquot_Submitter_ID_cor"
# Merge "sample_of_PDC_for_TCGA" with "PDC_biospecimen_manifest_distinct" to get clinical information for each sample.
sample_of_PDC_for_TCGA_with_biospecimen_manifest <- inner_join(
sample_of_PDC_for_TCGA,
PDC_biospecimen_manifest_distinct,
by = "Case_ID",
suffix = c("_sample_of_PDC_for_TCGA", "_PDC_biospecimen_manifest_distinct"))
# by = "Case_ID",
# by = "Aliquot_Submitter_ID_cor",
# by = join_by(Case_ID, Aliquot_Submitter_ID_cor),
# I think inner_join() by = join_by(Case_ID, Aliquot_Submitter_ID_cor), is more reliable for case matching.
dim(sample_of_PDC_for_TCGA_with_biospecimen_manifest)
colnames(sample_of_PDC_for_TCGA_with_biospecimen_manifest)ここで、遺伝子発現プロファイルを読み込む。使用したデータは以前ダウンロードしたものである。TCGAは一回のダウンロードにつき10000ファイルしかダウンロード出来ないので、3回に分けてダウンロードしている。なので、biospeciemenも3つ出来る。データフレームbiospeciemen_GDC_1、biospeciemen_GDC_2、biospeciemen_GDC_3として読み込んできて、それらをリストtempにいれ、縦方向(行方向)に結合することでデータフレームbiospeciemen_GDCを作成する。上記と同様、そのデータフレームbiospeciemen_GDCの列名に含まれているスペースをアンダーバーに置換しておく。そして、ここでも列Case_IDの値の重複を除いておき、それをデータフレームbiospeciemen_GDC_disitict_Case_IDとする。
# code 65
# BE CAREFULL!!!!
# THIS IS READ COUNT DATA OF TRANSCRIPTOME FROM GDC PORTAL, NOT PDC PORTAL
biospeciemen_GDC_1 <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_sample_sheet.2025-10-25.tsv")
biospeciemen_GDC_2 <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_sample_sheet.2025-10-25 (1).tsv")
biospeciemen_GDC_3 <- read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_sample_sheet.2025-10-25.tsv")
temp <- list()
temp[[1]] <- biospeciemen_GDC_1
temp[[2]] <- biospeciemen_GDC_2
temp[[3]] <- biospeciemen_GDC_3
biospeciemen_GDC <- reduce(temp, rbind)
# Replace " " into "_" in columns of "biospeciemen_GDC".
colnames_biospeciemen_GDC <- gsub(pattern = " ", replacement = "_", x = colnames(biospeciemen_GDC))
colnames(biospeciemen_GDC) <- colnames_biospeciemen_GDC
# biospeciemen_GDC <- read_tsv(
# file = "C:/Users/KatsutoshiSato/Documents/Data/KS20240724TR B049 0010 19/CPTAC_RNAseq_GDC_assosiated_file/gdc_sample_sheet.2024-07-31.tsv"
# )
biospeciemen_GDC_disitict_Case_ID <- biospeciemen_GDC %>% dplyr::distinct(Case_ID, .keep_all = TRUE)
biospeciemen_GDC_disitict_Case_ID_rename <- biospeciemen_GDC_disitict_Case_ID %>% rename("Case_Submitter_ID" = "Case_ID")データフレームsample_of_PDC_for_TCGA_with_biospecimen_manifest(相関の解析に使用出来そうなタンパク質発現プロファイルの列名とそれに一致する試料の情報)とデータフレームbiospeciemen_GDC_disitict_Case_ID_rename(遺伝子発現プロファイルで使用された試料の情報)は、それぞれ既に不要な重複が解消されているので、それを列Case_Submitter_IDでinner_join()し、データフレームCase_matching_between_PDC_and_GDCを作成する。そしてこのデータフレームCase_matching_between_PDC_and_GDCをTSVファイルとして外部に保存する。
# code 66
# Merge following 2 datasets.
# "sample_of_PDC_for_TCGA_with_biospecimen_manifest" which is the file name of mass spec count data with Case ID, and
# "biospeciemen_GDC_disitict_Case_ID_rename" which is GDC sample sheet.
# "sample_of_PDC_for_TCGA_with_biospecimen_manifest" IS DATASET OF PDC PORTAL, NOT GDC PORTAL.
Case_matching_between_PDC_and_GDC <- inner_join(
sample_of_PDC_for_TCGA_with_biospecimen_manifest,
biospeciemen_GDC_disitict_Case_ID_rename,
by = "Case_Submitter_ID",
suffix = c("_sample_of_PDC_for_TCGA_with_biospecimen_manifest", "_biospeciemen_GDC_disitict_Case_ID_rename"))
dim(Case_matching_between_PDC_and_GDC)
colnames(Case_matching_between_PDC_and_GDC)
# Check the below using "biospeciemen_cor" and "file_name_case_id_cor"
# Case_matching_between_PDC_and_GDC$Case_ID == "9a58eb4b-63d7-11e8-bcf1-0a2705229b82"
# Case_matching_between_PDC_and_GDC$name == "PDC000113_25_1312_01A_Log_Ratio"
# Case_matching_between_PDC_and_GDC$Aliquot_Submitter_ID_cor_sample_of_PDC_for_TCGA == "25_1312_01A"
write_tsv(Case_matching_between_PDC_and_GDC, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/Case_matching_between_PDC_and_GDC.tsv")
ここまでの解析をRDataとして外部に保存する。
# code 67
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/2026 01 10 CPTAC expression matrix_4.RData")一応、解析環境も出しておく。
# code 68
sessioninfo::session_info()ここから使用するRのダウンロード3
ここから、Rのコードを変える。上記までを一旦閉じて、新しく以下のRを開く。コードはここで販売している。ZIPを解凍するとRのコードがいくつかあるので、そのうちの「3 case matching of GDC and PDC.Rmd」というファイルが、以降のコードである。
ここでは、遺伝子発現プロファイル名を読み込んで、ファイル名(これが遺伝子発現プロファイル名)から成るベクトルである。これらはは以前ダウンロードしたものである。
# case 69
GDC_tumor <- list.files(path = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor")
GDC_normal <- list.files(path = "/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal") そして、上記code 66で出力しておいたCase_maching_between_PDC_and_GDC.tsvを読み込んで、それをデータフレームmatching_GDC_PDCとする。
# case 70
matching_GDC_PDC <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/Case_maching_between_PDC_and_GDC.tsv") データフレームmatching_GDC_PDCの列File_IDの値のうち、一致するTCGAのファイル名と一致する値を取得する。TCGAので遺伝子発現プロファイルはがん組織と正常組織で分けて作成しているので、そのそれぞれを使って、データフレームmatching_GDC_PDCから一致する列File_IDを抽出してきて、ベクトルGDC_for_correlation_normal、ベクトルGDC_for_correlation_tumorを作成する。
# case 71
GDC_for_correlation_normal <- matching_GDC_PDC[matching_GDC_PDC$File_ID %in% GDC_normal,]$File_ID
view(GDC_for_correlation_normal)
GDC_for_correlation_tumor <- matching_GDC_PDC[matching_GDC_PDC$File_ID %in% GDC_tumor,]$File_ID
view(GDC_for_correlation_tumor)
上記で作成したファイル名が記載されたベクトルGDC_for_correlation_normal、ベクトルGDC_for_correlation_tumorと、ファイルが保存されたディレクトリのパスを結合し、ベクトルGDC_count_normal、ベクトルGDC_count_tumorを作成する。これが、これまでに選んできたタンパク質発現プロファイルと同じ試料から得られた遺伝子発現プロファイルのファイルパスになる。
# case 72
GDC_count_normal <- paste0("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_normal/", GDC_for_correlation_normal)
GDC_count_tumor <- paste0("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/count_tumor/", GDC_for_correlation_tumor)
view(GDC_count_normal)
view(GDC_count_tumor)上記で作成したファイルパスのベクトルを使って、遺伝子発現プロファイルを、この解析用のディレクトリにコピーする。これで、相関解析に使用するTCGAの遺伝子発現プロファイルを選んでくることが出来る。
# case 73
# Done at 2026 01 31
file.copy(from = GDC_count_normal,
to = "/mnt/seqdata/public_data/Blog/2026 01 03 CPTAC/RNAseq_matching_GDC_PDC/", overwrite = TRUE, recursive = TRUE,
copy.mode = TRUE, copy.date = FALSE)
file.copy(from = GDC_count_tumor,
to = "/mnt/seqdata/public_data/Blog/2026 01 03 CPTAC/RNAseq_matching_GDC_PDC/", overwrite = TRUE, recursive = TRUE,
copy.mode = TRUE, copy.date = FALSE)ここから使用するRのダウンロード4
ここから、Rのコードを変える。上記までを一旦閉じて、新しく以下のRを開く。コードはここで販売している。ZIPを解凍するとRのコードがいくつかあるので、そのうちの「4 GDC expression matrix.Rmd」というファイルが、以降のコードである。
そして、改めてそれらのファイルパスをベクトルGDCにいれ、その中からファイルの末尾がcounts.tsvで終わっているものをベクトルGDC_countとする。
# case 74
GDC <- list.files("/mnt/seqdata/public_data/Blog/2026 01 03 CPTAC/RNAseq_matching_GDC_PDC", all.files = TRUE, full.names = TRUE, recursive = TRUE)
GDC_count <- GDC[str_detect(pattern = "counts.tsv$", string = GDC)]そのファイルパスを保持したベクトルを使って、それぞれのファイルを読み込んできて、リストGDC_count_collectionの各要素に入れいき、各要素に入っているデータフレームの不要な列を除く。
# case 75
# GDC_count_collection<- list()
#
#
# # This is test.
# foreach(i = 1:10, .packages = 'readr', .combine = 'list') %do% {
# GDC_count_collection[[i]] <- read.table(
# file = GDC_count[i],
# sep = "\t",
# header = TRUE)
# GDC_count_collebction[[i]] <- GDC_count_collection[[i]][-c(1,2,3,4),]
# } # It looks good.
# # This is test.
# foreach(i = 1:ength(GDC_count), .packages = 'readr', .combine = 'list') %do% {
# GDC_count_collection[[i]] <- read.table(
# file = GDC_count[i],
# sep = "\t",
# header = TRUE)
# GDC_count_collebction[[i]] <- GDC_count_collection[[i]][-c(1,2,3,4),]
# } # It is probably right code, but calculation does not start for some reason.
# for (i in 1:length(GDC_count)) {
# GDC_count_collection[[i]] <- read_tsv(GDC_count[i])} # this is too long to read table.
GDC_count_collection<- list()
for (i in 1:length(GDC_count)) {
GDC_count_collection[[i]] <- read.table(
file = GDC_count[i],
sep = "\t",
header = TRUE)
GDC_count_collection[[i]] <- GDC_count_collection[[i]][-c(1,2,3,4),]
GDC_count_collection[[i]] <- GDC_count_collection[[i]][,c(1, 2, 7)]
} # This takes ~10 min to read it.
#GDC_count_collection[[i]] <- GDC_count_collection[[i]][,c(1, 2, 7)]
# GDC_count_collection_unstrand<- list()
# for (i in 1:length(GDC_count_collection)) {
# GDC_count_collection_unstrand[[i]] <- GDC_count_collection[[i]][,c(2, 4)]
# } # This takes ~10 min to read it.
ベクトルGDC_countには、遺伝子発現プロファイルのあるパスが入っている。その文字列のうち、最初のスラッシュまでの文字列を削除(何もなしで置き換え)し、それをデータフレームGDC_file_nameとする。
# case 76
# extract_between_slash_and_dot <- function(file_path) {
# after_last_slash <- sub("^.*/", "", file_path)
# extracted_string <- sub("\\..*$", "", after_last_slash)
# return(extracted_string)
# }
#
# GDC_file_name <- sapply(GDC_count, extract_between_slash_and_dot) %>% data.frame()
# GDC_file_name <- GDC_file_name %>% rename("file_name" = ".")
# view(GDC_file_name)
GDC_file_name <- sub("^.*/", "" , GDC_count) %>% data.frame()
view(GDC_file_name)データフレームGDC_file_nameに入っている値を使って、リストGDC_count_collectionの各要素のデータフレームの各列に、列名を付ける。1列目がgene_id、2列めがgene_name、3列目がcode 76で作成したファイル名である。
# case 77
for (i in 1:length(GDC_count_collection)) {
temp <- c("gene_id","gene_name", GDC_file_name[i,])
colnames(GDC_count_collection[[i]]) <- temp
}リストGDC_count_collectionの各要素をinner_join()して、横方向(列方向)に結合させ、データフレームGDC_count_mergeを作成する。
# case 78
GDC_count_merge <- purrr::reduce(GDC_count_collection, inner_join)
# test_merge <- GDC_count_collection[[1]] %>% inner_join(GDC_count_collection[[2]], keep = FALSE)
データフレームGDC_count_mergeの列gene_idを行名にして、列gene_nameを削除し、そのデータフレームをマトリックスGDC_count_merge_matに変換する。
# case 79
GDC_count_merge_mat <- GDC_count_merge %>% column_to_rownames("gene_id") %>% dplyr::select(-one_of("gene_name")) %>% as.matrix()マトリックスGDC_count_merge_matの各遺伝子の発現量(カウント値)がゼロだった症例の数をカウントし、その数を列count_zeroに入れる。そして、全症例のうち5%以上で発現量がゼロだった遺伝子をデータフレームcount_zero_in_5_percent_of_patientとして、その遺伝子名をrownames()で取得し、ベクトルgenes_will_be_removedとする。
# case 80
count_zero <- apply(GDC_count_merge_mat, 1, function(x) sum(x == 0)) %>% data.frame()
colnames(count_zero) <- "number_of_patients_with_zero_count"
ncol(GDC_count_merge_mat) * 0.05 # 100.5 # 5% of patients have zero count.
count_zero_in_5_percent_of_patient <- count_zero %>% filter(number_of_patients_with_zero_count > ncol(GDC_count_merge_mat) * 0.05)
genes_will_be_removed <- rownames(count_zero_in_5_percent_of_patient)ここまでの結果をRDataとして外部に保存しておく。
# case 81
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/gdc expression matrix_1.RData")データフレームGDC_count_mergeから、ベクトルgenes_will_be_removedと一致する遺伝子、すなわち、全症例の5%以上の症例で発現量が確認できなかった遺伝子を除き、データフレームGDC_count_merge_remove_5_pct_zeroとする。そのデータフレームGDC_count_merge_remove_5_pct_zeroの列gene_idと列gene_nameをTSVファイルとして外部に保存しておく。
# case 82
# remove row count genes from dataset.
GDC_count_merge_remove_5_pct_zero <- GDC_count_merge %>% filter(!gene_id %in% genes_will_be_removed) # 21643
# write_tsv(GDC_count_merge_remove_5_pct_zero[,c(1,2)], "/media/kats/fimecs/KS20240724TR B049 0010 19/duplicated genes.tsv") # this file can be used for which genes are duplicated. However, I can not find duplicated genes,
write_tsv(GDC_count_merge_remove_5_pct_zero[,c(1,2)], "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/duplicated genes.tsv")ここで、遺伝子名が重複している遺伝子についてどうするか検討する。データフレームGDC_count_merge_remove_5_pct_zeroから、重複している遺伝子の取得し、それをデータフレームcheck_duplicated_genesとする。これらは異なる遺伝子IDが付けられている同じ遺伝子である。要は別々にアノテーションされてしまっていたり、何かしらのバリアントかもしれないし、もしかしたら構造がかなり似ている別物かもしれない遺伝子である。余談だが、RNA-seqの解析はこういう事があるので、Ensemble IDを行名にして解析していき、必要に応じて遺伝子名に直して解析を進める必要がある。
しかしながら、ここではどちらの遺伝子を使っていくか決める必要がある。タンパク質と遺伝子発現量の相関を解析する必要がある。そのために、上記で作成したデータフレームcheck_duplicated_genes(重複している遺伝子のみからなるデータフレーム)の列gene_nameを使って、まずはデータフレームGDC_count_merge_remove_5_pct_zero(全症例のうち5%以上の症例で発現量が確認できなかった遺伝子を除いたデータフレーム)から、重複している遺伝子のみの遺伝子発現プロファイルを取ってきて、それをデータフレームGDC_count_merge_remove_5_pct_zero_duplicatedとする。
….ちょっと待て。データフレームcheck_duplicated_genesとデータフレームGDC_count_merge_remove_5_pct_zero_duplicatedって、同じものじゃあないか…やってしまったわ。ま、いいか。
データフレームGDC_count_merge_remove_5_pct_zero_duplicatedの3列目以降(すなわち列gene_idと列gene_name以外の各疾患の遺伝子発現量)のrowSums()を計算して、その結果を列sum_countに入れる。rowSums()は、行の和、つまりここでは、ある遺伝子の症例全体の総和である。そして、そして、その列sum_countの値が、多い方の遺伝子を取ってきて、それをデータフレームGDC_count_merge_remove_5_pct_zero_duplicated_2とする。
このデータフレームGDC_count_merge_remove_5_pct_zero_duplicated_2をデータフレームGDC_count_merge_remove_5_pct_zeroに加えるため、まずはデータフレームGDC_count_merge_remove_5_pct_zeroから、重複のある遺伝子すべてを除去してデータフレームGDC_count_merge_remove_5_pct_zero_remove_duplicatedを作成し、それとデータフレームGDC_count_merge_remove_5_pct_zero_duplicated_2をbind_rows()し、データフレームGDC_count_merge_remove_5_pct_zero_remove_duplicatedを作成する。これで、重複した遺伝子のうち、総カウント数が多い方の遺伝子を残すことが出来る。
# case 83
# count duplicated gene name
table(GDC_count_merge_remove_5_pct_zero$gene_name) %>% data.frame() %>% filter(Freq > 1) # check number of duplication
check_duplicated_genes <- GDC_count_merge_remove_5_pct_zero %>% group_by(gene_name) %>% filter(n() > 1) # extract duplicated gene names.
GDC_count_merge_remove_5_pct_zero_duplicated <- GDC_count_merge_remove_5_pct_zero[
GDC_count_merge_remove_5_pct_zero$gene_name %in% check_duplicated_genes$gene_name, ] # extract duplicated genes with count. This is same as the above.
# rowSums
GDC_count_merge_remove_5_pct_zero_duplicated$sum_count <- rowSums(GDC_count_merge_remove_5_pct_zero_duplicated[,3:ncol(GDC_count_merge_remove_5_pct_zero_duplicated)])
# remove duplicated genes.
GDC_count_merge_remove_5_pct_zero_duplicated_2 <- GDC_count_merge_remove_5_pct_zero_duplicated %>%
group_by(gene_name) %>%
filter(sum_count == max(sum_count))
# remove duplicated genes from the dataset without gene count with zero count less than 5%
GDC_count_merge_remove_5_pct_zero_remove_duplicated <- GDC_count_merge_remove_5_pct_zero[GDC_count_merge_remove_5_pct_zero$gene_id %in% check_duplicated_genes$gene_id == FALSE, ]
# remove duplicated genes and combine the dataset.
GDC_count_merge_remove_5_pct_zero_remove_duplicated <- bind_rows(GDC_count_merge_remove_5_pct_zero_remove_duplicated, GDC_count_merge_remove_5_pct_zero_duplicated_2)データフレームGDC_count_merge_remove_5_pct_zero_remove_duplicatedから列gene_idを除き、列gene_nameをcolumn_to_rownames()で行名にし、それをデータフレームGDC_count_merge_remove_5_pct_zero_remove_duplicated_2とする。それを転置させて、データフレームGDC_count_merge_remove_5_pct_zero_remove_duplicated_2_tとする。その行名と列File Nameにし、さらにそれを列File_Nameに直す。これを今後使用していく。そして、データフレームGDC_count_merge_remove_5_pct_zero_remove_duplicated_2は、行名を列Geneにして、TSVファイルで外部に保存しておく。
# case 84
GDC_count_merge_remove_5_pct_zero_remove_duplicated_2 <- GDC_count_merge_remove_5_pct_zero_remove_duplicated %>% select(-one_of("gene_id")) %>% column_to_rownames("gene_name") # Need to be removed duplicated genes.
GDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t <- GDC_count_merge_remove_5_pct_zero_remove_duplicated_2 %>% t() %>% as.data.frame()
GDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t <- GDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t %>% rownames_to_column(var = "File Name")
GDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t <- GDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t %>% rename("File_Name" = "File Name")
gdc <- GDC_count_merge_remove_5_pct_zero_remove_duplicated_2 %>% rownames_to_column(var = "Gene")
write_tsv(gdc, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/gdc.tsv")ここまでの結果をRDataとして外部に保存しておく。
# case 85
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/gdc expression matrix_2.RData")次に、TGCAの遺伝子発現プロファイルの取得に用いた試料の情報が書かれているTSVファイルであるgdc_sample_sheet.tsvを読んでくる。これも以前の記事でダウンロードしたものである。これも上述と同様で、3つに分割してダウンロードしているので、読み込んだらそれをリストtempに入れて、それをreduce()でbind_rows()して縦方向(行方向)に結合させておく。それをデータフレームGDC_sample_sheetとする。そして、列名に入っている邪魔くさいスペースをアンダーバーに置き換えておく。
# case 86
temp <- list()
temp[[1]] <- readr::read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/1_ffp_frozen_oct/gdc_sample_sheet.2025-10-25.tsv")
temp[[2]] <- readr::read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/2_snapfrozen_unkown/gdc_sample_sheet.2025-10-25 (1).tsv")
temp[[3]] <- readr::read_tsv("/mnt/seqdata/public_data/Blog/2025 10 18 TCGA/3_normal/gdc_sample_sheet.2025-10-25.tsv")
GDC_sample_sheet <- reduce(temp, bind_rows)
GDC_sample_sheet_colnames <- gsub(pattern = " ", replacement = "_", x = colnames(GDC_sample_sheet))
colnames(GDC_sample_sheet) <- GDC_sample_sheet_colnamesそして、各症例の遺伝子発現プロファイルであるデータフレームGDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t(全症例のうち5%以上の症例で発現量が確認できなかった症例を除き、遺伝子の重複を解消したデータフレーム)と、遺伝子発現プロファイルの取得に使用した試料の情報をまとめたデータフレームGDC_sample_sheetを列FIle_Nameを軸にしてinner_join()し。データフレームGDC_sample_sheet_w_countを作成する。そのデータフレームの列Case_IDの値のうち、「,」から最後までの文字列を削除しておく。確かに、これらは正常組織とがん組織とか、謎試料のプールとかだったはずである。さすがTCGA、運用が長いだけあって、遺伝子発現プロファイルと症例を関連付けるのがすごく楽….
# case 87
GDC_sample_sheet_w_count <- GDC_sample_sheet %>% dplyr::inner_join(GDC_count_merge_remove_5_pct_zero_remove_duplicated_2_t, by = "File_Name")
GDC_sample_sheet_w_count$Case_ID <- sub(pattern = ",.*$", "", GDC_sample_sheet_w_count$Case_ID)
出来上がったデータフレームGDC_sample_sheet_w_countはTSVファイルとして外部に保存しておく。
# case 88
write_tsv(GDC_sample_sheet_w_count, "/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/gdc_sample_sheet_w_count.tsv")ここまでをRDataとして外部に保存しておく。
# case 89
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/gdc expression matrix_3.RData")
sessionInfo()ここから使用するRのダウンロード5
ここから、Rのコードを変える。上記までを一旦閉じて、新しく以下のRを開く。コードはここで販売している。ZIPを解凍するとRのコードがいくつかあるので、そのうちの「5 Correlation between PDC and GDC.Rmd」というファイルが、以降のコードである。
これまで整えてきたデータフレームはTSVファイルで外部に保存されているはずなので、それらを読み込んでいく。もやはどのデータは何なのかすっかり忘れているので、以下の表にまとめておく。
| 元のデータフレーム名 | 作成したコード | ここでのデータフレーム名 | 内容 |
|---|---|---|---|
| result_missForest_post_df_matched_case_ID | code 63 | pdc | missForestで欠損値を補完した後、TCGAの症例と一致させた症例のタンパク質発現プロファイル |
| gdc_sample_sheet_w_count | code 87 | gdc | CPTACと症例を一致させたTCGAの遺伝子発現プロファイル |
| Case_maching_between_PDC_and_GDC | code 66 | case_matching | 相関の解析に使用出来そうなタンパク質発現プロファイルの列名とそれらの試料の情報と、遺伝子発現プロファイルの試料の情報を一致させたデータ |
読み込んだら、データフレームcase_matchingの列File_IDのうち、データフレームgdcの列File_IDが一致しているものを抽出してきてデータフレームcase_matching_2とする。このデータフレームcase_matching_2の列nameには、タンパク質発現プロファイル名が入っているのだが、その値の最初にPDCから始まる番号とアンダーバーが、例えばPDC000110_という具合に付いている。それをseparate()で最初のアンダーバーとそれ以降を分け、列nameとし、それをデータフレームcase_matching_2_wo_PDC_Study_IDとする。また、データフレームpdcの列名も同様に頭のPDCから始まる番号を最初のアンダーバーで分け、列名を直しておく。そしてデータフレームpdcの列、すなわち各症例のタンパク質発現プロファイルのうち、データフレームcase_matching_2_wo_PDC_Study_IDの列nameに一致する列を選んできて、それをデータフレームpdc_2とする。このデータフレームpdc_2の列proteinをcolumn_to_rownames()で行名にし、それをデータフレームpdc_3とする。このデータフレームpdc_3を転置させてデータフレームpdc_4を作成し、さらにrownames_to_column()で列名を列nameに直し、それをデータフレームpdc_5とする。確認しやすくするためにデータフレームpdc_5は列nameで並べ替えておき、またその列nameを行名に直し、データフレームpdc_6とする。念の為、タンパク質に重複がないか53行目から55行目で確認する。
続いてデータフレームgdcを整理する。データフレームgdcとタンパク質発現プロファイルに関連した試料の情報を結びつけたい。そのときに必要になるのが、既に一致する試料の情報が揃っているデータフレームcase_matching_2_wo_PDC_Study_IDであるが、こんなに沢山の列は要らないので、必要な列である列nameと列File_IDを選んできて、データフレームcase_matching_3とする。それと、データフレームgdcをinner_join()すると、タンパク質発現プロファイル名と遺伝子発現プロファイル名を結びつける事が出来る。それをデータフレームgdc_2とする。ここで、データフレームgdc_2の列nameがNAでないものを抽出してデータフレームgdc_3を作っているが、それは実際には不要である。解析の途中のチェックの名残である。別に有っても影響はないので、このまま進める。また、その下の79行目から80行目にはデータフレームgdc_3の列nameの重複が確認しているが、これも確認のためである。80行目では、重複している遺伝子を除いてデータフレームgdc_4を作成しているが、そもそも重複していなかったので、これも解析中の名残である。データフレームgdc_4から、各遺伝子の症例ごとの発現量を選んできて、それをデータフレームgdc_5とし、それを、タンパク質発現プロファイル名を1列目に、2列目から遺伝子発現量になるように並べ替え、データフレームgdc_6とする。このデータフレームgdc_6も列nameで並べ替え、それをデータフレームgdc_7に、さらにcolumn_to_rownames()で列nameを行名にして、それをデータフレームgdc_8にする。そして、データフレームgdc_8の列のうち、データフレームpdc_6の列と一致するものを選んできて、それをデータフレームgdc_9とする。このデータフレームgdc_9も念のため遺伝子に重複がないことを確認してく。また、データフレームpdc_6の列のうち、データフレームgdc_6の列と一致しするものも選んできて、それをデータフレームpdc_7とする。107行目から126行目までは、データフレームpdc_7とデータフレームgdc_9のそれぞれタンパク質、遺伝子が、同じ順番になるように並べ替えて、両者が同じであることを確認している。
このあたりの処理は%>%で一気につなげることも出来そうだが、このときは様子を見ながら随分と対話的に解析して行ったので、こんなに細切れになってしまっている。また、過剰に症例、遺伝子、タンパク質が一致しているのかを確かめているのは、エラーがあったら逐一戻ったほうが、要はトラブルがないか確認しながら処理を進めた方が、最終的に楽だからである。
# case 90
pdc <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/result_missForest_pre_argu_df_median_normalized_df_matched_case_ID_output.tsv")
gdc <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/gdc_sample_sheet_w_count.tsv")
case_matching <- read_tsv("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/Case_matching_between_PDC_and_GDC.tsv")
# case_matching$`File ID`
# gdc$`File ID`
case_matching_2 <- case_matching[case_matching$File_ID %in% gdc$File_ID == TRUE,]
case_matching_2_wo_PDC_Study_ID <- case_matching_2 %>% separate(name, into = c("PDC_Study_ID", "name"), sep = "_", extra = "merge")
colnames_pdc <- colnames(pdc) %>% data.frame() %>% rename("name" = ".") %>% separate(name, into = c("PDC_Study_ID", "name"), sep = "_", extra = "merge")
colnames_pdc[1,2] <- "protein"
colnames(pdc) <- colnames_pdc$name
check <- case_matching_2_wo_PDC_Study_ID$name
length(check) # 2176
view(check)
check_2 <- colnames(pdc[,2:ncol(pdc)])
length(check_2) # 3248
view(check_2)
table(check %in% check_2)
# TRUE
# 2176
pdc_2 <- pdc[,c(1, which(colnames(pdc) %in% case_matching_2_wo_PDC_Study_ID$name))]
ncol(pdc_2) # 2176 cases + Gene = 2177
# To leave column name and row names as is, set column "Gene" as row names.
pdc_3 <- pdc_2 %>% column_to_rownames(var = "protein")
# transpose dataframe pdc_4
pdc_4 <- as.data.frame(t(pdc_3))
# Get back row names to colums to sort them
pdc_5 <- pdc_4 %>% rownames_to_column(var = "name")
pdc_5 <- arrange(pdc_5, name)
# again set column "name" as row name.
pdc_6 <- pdc_5 %>% column_to_rownames(var = "name")
# dimension of pdc_6
dim(pdc_6) # cases, genes = 2176 5208
proteins <- colnames(pdc_6)
check_protein <- table(proteins) %>% data.frame() %>% filter(Freq >1) # There is no duplicated protein.
# case_matching$`File ID`
# gdc$`File ID`
# Extract just only the requred columns
case_matching_3 <- case_matching_2_wo_PDC_Study_ID %>% select(name, File_ID)
# Following is better than the follow;
# gdc_2 <- gdc[gdc$`File ID` %in% case_matching_2$`File ID` == TRUE,]
# Because other information are able to be retrieved.
nrow(gdc) # 1484
nrow(case_matching_3) # 2176
gdc_2 <- inner_join(gdc, case_matching_3)
nrow(gdc_2) # 2176
gdc_3 <- gdc_2 %>% filter(name != "NA")
nrow(gdc_3) # 2176
# check duplicated "name"
check_gdc_3_duplicated_name <- table(gdc_3$name) %>% data.frame() %>% rename("name" = "Var1") %>% filter(Freq > 1)
gdc_4 <- gdc_3[gdc_3$name %in% check_gdc_3_duplicated_name$name == FALSE,]
gdc_5 <- gdc_4[,12:ncol(gdc_4)]
gdc_6 <- gdc_5 %>% select(c("name", colnames(gdc_5[1:ncol(gdc_5)-1])))
gdc_7 <- arrange(gdc_6, name)
gdc_8 <- gdc_7 %>% column_to_rownames(var = "name")
# dimension of gdc_8
dim(gdc_8) # cases, genes = 2176 20749
gdc_9 <- gdc_8[, colnames(gdc_8) %in% colnames(pdc_6) == TRUE]
# dimension of gdc_9
dim(gdc_9) # cases, genes = 2176 5130
genes <- colnames(gdc_9)
check_genes <- table(genes) %>% data.frame() %>% filter(Freq >1) # There is no duplicated genes
# the gene number in gdc_9 (5127) is less than the protein numnber in pdc_6 (5208).
# Therefore, match protein number of pdc and gene number of gdc.
pdc_7 <- pdc_6[, colnames(pdc_6) %in% colnames(gdc_9) == TRUE]
# dimension of pdc_7
dim(pdc_7) # cases, genes = 2176 5130
# sort protein name of pdc_7
protein_name_2 <- data.frame(protein = colnames(pdc_7))
protein_name_2 <- protein_name_2 %>% arrange(protein)
pdc_7 <- pdc_7 %>% select(all_of(protein_name_2$protein))
# sort protein name of gdc_6
gene_name_2 <- data.frame(gene = colnames(gdc_9))
gene_name_2 <- gene_name_2 %>% arrange(gene)
gdc_9 <- gdc_9 %>% select(all_of(gene_name_2$gene))
# check order of genes and proteins
all(colnames(pdc_7) == colnames(gdc_9)) # TRUE
# double check whether the order of protein_name_2 and gene_name_2 are same.
order <- data.frame(collect = rep(NA, nrow(protein_name_2)))
for(i in 1:nrow(protein_name_2)){
order$collect[i] <- protein_name_2[i,1] == gene_name_2[i,1]
}
table(order$collect)
# TRUE
# 5130
ようやく、タンパク質発現量と遺伝子発現量の相関を求める。まず、リストcorrelationの各要素にデータフレームgdc_9とデータフレームpdc_7を入れ、各要素にそれぞれgeneとproteinと名前を付ける。また、結果を入れるためのデータフレームinterestを用意し、ピアソンの相関係数とそのp値、スピアマンの順位相関係数とそのp値、ケンドールの順位相関係数とそのp値を入れるための空の列を用意する。相関係数にはcor.test()を使って、引数で計算方法(ピアソン、スピアマン、ケンドール)を指定し、それぞれのデータフレームに入れていく。
一応、ちゃんと計算できるか確認するため、1列目だけを使ってケンドールの順位相関係数を計算して、データフレームtestにその結果を入れてみる。それで大丈夫なので、for(){}で一気に計算してしまう。リストの要素1000個毎にgc()で不要なデータを削除しておいた。
これだけ下準備に時間をかけた上で、必要な数字を出すのは一発で終わるっていうね…非常に複雑な気持ちである。
# case 91
# correlation with stats.
# gdc_9
# pdc_7
correlation <- list()
correlation[[1]] <- gdc_9 #%>% apply(MARGIN = 2, as.numeric)
correlation[[2]] <- pdc_7 #%>% apply(MARGIN = 2, as.numeric)
names(correlation) <- c("gene", "protein")
interest <- data.frame(gene_name = colnames(gdc_6))
interest$pearson <- rep(NA, nrow(interest))
interest$pearson_p <- rep(NA, nrow(interest))
interest$spearman <- rep(NA, nrow(interest))
interest$spearman_p <- rep(NA, nrow(interest))
interest$kendall <- rep(NA, nrow(interest))
interest$kendall_p <- rep(NA, nrow(interest))
test <- cor.test(correlation$gene[,1], correlation$protein[,1], method = "kendall")
for(i in 1:length(correlation[[1]])){
#
print(paste0("Now processing ", i, " of " , length(correlation[[1]])))
#
interest$pearson[i] <- cor.test(correlation$gene[,i], correlation$protein[,i], method = "pearson")[["estimate"]][["cor"]]
interest$pearson_p[i] <- cor.test(correlation$gene[,i], correlation$protein[,i], method = "pearson")[["p.value"]]
interest$spearman[i] <- cor.test(correlation$gene[,i], correlation$protein[,i], method = "spearman")[["estimate"]][["rho"]]
interest$spearman_p[i] <- cor.test(correlation$gene[,i], correlation$protein[,i], method = "spearman")[["p.value"]]
interest$kendall[i] <- cor.test(correlation$gene[,i], correlation$protein[,i], method = "kendall")[["estimate"]][["tau"]]
interest$kendall_p[i] <- cor.test(correlation$gene[,i], correlation$protein[,i], method = "kendall")[["p.value"]]
if(i == 1000){
gc(verbose = TRUE, reset = TRUE, full = TRUE)
gc(verbose = TRUE, reset = TRUE, full = TRUE)}
else if(i == 2000){
gc(verbose = TRUE, reset = TRUE, full = TRUE)
gc(verbose = TRUE, reset = TRUE, full = TRUE)}
else if(i == 3000){
gc(verbose = TRUE, reset = TRUE, full = TRUE)
gc(verbose = TRUE, reset = TRUE, full = TRUE)}
else if(i == 4000){
gc(verbose = TRUE, reset = TRUE, full = TRUE)
gc(verbose = TRUE, reset = TRUE, full = TRUE)}
else if(i == 5000){
gc(verbose = TRUE, reset = TRUE, full = TRUE)
gc(verbose = TRUE, reset = TRUE, full = TRUE)}
}データフレームinterestの各列にはそれぞれの計算結果が入っているが、相関係数はどのような分布になっているのかヒストグラムで見る。以下はピアソンの相関係数。
# case 92
hist(interest$pearson, breaks = 250, main = "Pearson", xlab = "Pearson correlation coefficient")次はスピアマン。
# case 93
hist(interest$spearman, breaks = 250, main = "Spearman", xlab = "Spearman correlation coefficient")次はケンドール。まぁ、みたところでわからん。わかることは、そんなに相関係数は高くないなってことである。相関係数は大体0.2ってところである。
# case 94
hist(interest$spearman, breaks = 250, main = "Kendall", xlab = "Kendall correlation coefficient")ここで、種々の細胞株のプロテオーム解析を行っている論文を見てみる。この論文のTable S4で、タンパク質と遺伝子発現量の相関が解析されているので、その値と比較してみる。まず、論文のサプリメントで公開されているエクセルのデータ(Table S4)をダウンロードして、そのエクセルファイルをパッケージopenxlsxのread.xlsx()で読み込んできて、データフレームPMID31978347_tableSとする。そうすると列Gene.Symbol、列Pearson、列Spearmanから成るデータフレームを読む事が出来る、それぞれ遺伝子、ピアソンの相関係数、スピアマンの順位相関係数の結果が入っている。それらの列名をそれぞれ列gene_name、列pearson_PMD31978347、列spearman_PMD31978347に変更し、上記で作成したデータフレームinterestとinner_join()して、データフレームinterest2を作成する。この論文は流石に細胞株を使っているだけあって、かなり相関が高い。0.7以上あるものまである。一方、この上記までで解析したものは精々が0.4くらいである。流石に、臨床検体で、しかも広く集めてきた試料ではこのくらいの相関係数なのかもしれない。結論としては、臨床検体をいかなるバイアスなしに解析したところで、その場合のタンパク質発現量と遺伝子発現量の相関は0.2くらいであって、きれいな相関を見るのは難しいということになる。
# Read table S4 of PMID: 31978347
# https://pubmed.ncbi.nlm.nih.gov/31978347/
# This paper is discribing correlation between gene and protein expression in CELL LINES!!
PMID31978347_tableS4 <- read.xlsx("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/Data PMID31978347/mmc4.xlsx",sheet = 2)
PMID31978347_tableS4 <- PMID31978347_tableS4 %>% rename("gene_name" = "Gene.Symbol",
"pearson_PMD31978347" = "Pearson",
"spearman_PMD31978347" = "Spearman")
interest_2 <- inner_join(interest, PMID31978347_tableS4, by = "gene_name")折角なので、論文とリポジトリ(CPTACとTCGA)に登録されている臨床検体の相関係数に相関があるのかを見てみる。散布図はplot()で、回帰直線はabline()で描く事が出来る。
結果、全く相関していない。少なくとも培養細胞と臨床検体は、全然違うってことだろう。
plot(x = interest_2$pearson, y = interest_2$pearson_PMD31978347,main = "Pearson's correlation")
abline(lm(interest_2$pearson_PMD31978347 ~ interest_2$pearson), col = "blue")
plot(x = interest_2$spearman, y = interest_2$spearman_PMD31978347,main = "Spearman's correlation")
abline(lm(interest_2$spearman_PMD31978347 ~ interest_2$spearman), col = "blue")そもそも遺伝子発現量とタンパク質発現量の解析は必要なのかで述べたが、自分でタンパク質発現と遺伝子発現の相関を解析しなくても、「おそらくこの論文で示されるくらいの相関はあるはずですよ」と結論しても良い気がする。しかし、その論文に関心のある遺伝子とタンパク質の相関が無いならば、もしかしたら自分で解析する羽目になるかもしれない。そんなときには、もしかしたら使えるかもしれない。
ここまでの結果をRDataで外部に保存しておく。
# case 96
save.image("/mnt/team4tb/Dropbox/Blog/2026 01 03 blog CPTAC/Correlation between PDC and GDC_1.RData")
sessionInfo()よくわからないタンパク質発現プロファイルの約90%が韓国と中国の研究機関発だった
code 46の32行目に書いてあるtable()の結果をみると、非常に興味深いことがわかる。各サンプルのタンパク質発現プロファイルと臨床情報を結びつけることが難しいかったデータの出所がデータフレームAliquot_Submitter_ID_2_difficult_to_be_matchedの列Project_Nameで見れるわけだが、504例中、35例がCPTAC3 Discovery and ConfirmatoryとProteogenomic Translational Research Centers (PTRC)で、残りの469例が韓国の研究機関だった。生存期間解析として使えなさそうなサンプルのなんと87%が韓国の研究機関由来ということになる。そして、これをよく見てみると、Aliquot Submitter IDが短い番号なので識別も難しいし、まだ理解できないのが、正常組織と腫瘍組織をプールして質量分析を行っているのもこのProject Nameである。プールしてもいいのだが、本来ならばタグを付けて区別がつくはずである。それにも関わらず、そのシグナルを分離していないのだろうか。自分が勉強不足の可能性あるのだが、普通はそこまで解析してからアップロードするのではないのだろうか。そうしなければ、解析するときに自分が困るのではないだろうか。
まず、研究における韓国の印象として、金の力を使って力技するけどその事業の見通しは甘いような感じがあり、これは自分が博士課程から思っていることである。最近でもたまに日本の学会でシンポジストだったりするが、そのときもわかりにくい発表をただ垂れ流しているようにしか見えない…この解析でもわかるようにトランスレーショナルスタディーも大規模にやっているようだったが、そんな印象を受けた。こういうことって、データの取り扱いや、その他の色々な場面にも当然ながら反映されるんだろう。要注意である。この国の科学は今のところ信頼は出来ないなって感じである…
中国は昨今では、勢いだけではアメリカを追い越す勢いであり、研究だけでなく産業についても日本は足元にも及んでいない。ただし、まだゴミだらけなのも確かなようだ。クオリティーはしっかりと見極める必要がある。
まとめ
以上、自分が行ったCPTACに登録されているプロテオームのデータを使った患者の生存期間の解析である。ほとんどデータの整理しかしていないという、なんとも面白くない解析だった。しかも、エンリッチメント解析、バイオリンプロット、生存解析など、重要なところは以前に行ったTCGAの解析をそのまま流用出来たので、何もやっていない感がある。逆に言ってみれば、一度しっかり解析を行ったら、そのコードを使いまわす事が出来るということではある。
ここではタンパク質発現量と遺伝子セットのエンリッチメントスコアが全生存期間に与える影響しか示さなかったが、がんのステージなどの階層も加えることが出来る。さらに、データフレームtreatmentで、抗がん剤投与や放射線照射の有無でも患者を分けることも出来る。
ただ、タンパク質発現量と遺伝子セットのエンリッチメントスコアをいちいちRで読んで、RDataをロードしてとかだと、面倒過ぎるので、なんとかしてShinyで表示出来るようにしておきたい。